
                   |
MOLCAS manual:
Next: List of Figures
Up: manual
Previous: 10.9 Core and Embedding Potentials
- 1
-
Björn O. Roos, Valera Veryazov, and Per-Olof Widmark.
Relativistic atomic natural orbital type basis sets for the alkaline
and alkaline-earth atoms applied to the ground-state potentials for the
corresponding dimers.
Theor. Chem. Acc., 111:345–351, 2004.
- 2
-
Björn O. Roos, Roland Lindh, Per-Åke Malmqvist, Valera Veryazov, and
Per-Olof Widmark.
Main group atoms and dimers studied with a new relativistic ANO
basis set.
J. Phys. Chem. A, 108:2851–2858, 2004.
- 3
-
Björn O. Roos, Roland Lindh, Per-Åke Malmqvist, Valera Veryazov, and
Per-Olof Widmark.
New relativistic ANO basis sets for transition metal atoms.
J. Phys. Chem. A, 109:6575–6579, 2005.
- 4
-
Björn O. Roos, Roland Lindh, Per-Åke Malmqvist, Valera Veryazov, and
Per-Olof Widmark.
New relativistic ANO basis sets for actinide atoms.
Chem. Phys. Letters, 409:295–299, 2005.
- 5
-
Björn O. Roos, Roland Lindh, Per-Åke Malmqvist, Valera Veryazov,
Per-Olof Widmark, and Antonio Carlos Borin.
New relativistic atomic natural orbital basis sets for lanthanide
atoms with applications to the 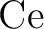 diatom and diatom and 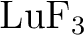 . .
J. Phys. Chem. A, 112:11431–11435, 2008.
- 6
-
Björn O. Roos, Per-Åke Malmqvist, and Laura Gagliardi.
Heavy element quantum chemistry – the multiconfigurational approach.
In Erkki J. Brändas and Eugene S. Kryachko, editors, Fundamental World of Quantum Chemistry. Vol. II, pages 425–442. Kluwer
Academic Publishers, Dordrecht, Netherlands, 2003.
- 7
-
Francesco Aquilante, Roland Lindh, and Thomas Bondo Pedersen.
Unbiased auxiliary basis sets for accurate two-electron integral
approximations.
J. Chem. Phys., 127:114107, 2007.
- 8
-
Francesco Aquilante, Per-Åke Malmqvist, Thomas Bondo Pedersen, Abhik Ghosh,
and Björn O. Roos.
Cholesky decomposition-based multiconfiguration second-order
perturbation theory (CD-CASPT2): Application to the spin-state energetics
of
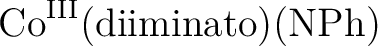 . .
J. Chem. Theory Comput., 4:694–702, 2008.
- 9
-
Francesco Aquilante, Thomas Bondo Pedersen, Björn O. Roos, Alfredo
Sánchez de Merás, and Henrik Koch.
Accurate ab initio density fitting for multiconfigurational
self-consistent field methods.
J. Chem. Phys., 129:024113, 2008.
- 10
-
Quan Manh Phung, Sebastian Wouters, and Kristine Pierloot.
Cumulant approximated second-order perturbation theory based on the
density matrix renormalization group for transition metal complexes: A
benchmark study.
J. Chem. Theory Comput., 12(9):4352–4361, 2016.
- 11
-
Sebastian Wouters, Veronique Van Speybroeck, and Dimitri Van Neck.
DMRG-CASPT2 study of the longitudinal static second
hyperpolarizability of all-trans polyenes.
J. Chem. Phys., 145(5):054120, 2016.
- 12
-
Naoki Nakatani and Sheng Guo.
Density matrix renormalization group (DMRG) method as a common tool
for large active-space CASSCF/CASPT2 calculations.
J. Chem. Phys., 146(9):094102, 2017.
- 13
-
Dongxia Ma, Giovanni Li Manni, and Laura Gagliardi.
The generalized active space concept in multiconfigurational
self-consistent field methods.
J. Chem. Phys., 135:044128, 2011.
- 14
-
Björn O. Roos.
The multiconfigurational (MC) self-consistent field (SCF) theory.
In Björn O. Roos, editor, Lecture Notes in Quantum
Chemistry. European Summer School in Quantum Chemistry, volume 58 of Lecture Notes in Chemistry, pages 177–254. Springer-Verlag, Berlin,
Germany, 1992.
- 15
-
James Finley, Per-Åke Malmqvist, Björn O. Roos, and Luis
Serrano-Andrés.
The multi-state CASPT2 method.
Chem. Phys. Letters, 288:299–306, 1998.
- 16
-
John D. Watts, Jürgen Gauss, and Rodney J. Bartlett.
Coupled-cluster methods with noniterative triple excitations for
restricted open-shell Hartree–Fock and other general single determinant
reference functions. Energies and analytical gradients.
J. Chem. Phys., 98:8718–8733, 1993.
- 17
-
Pavel Neogrády and Miroslav Urban.
Spin-adapted restricted Hartree–Fock reference coupled-cluster
theory for open-shell systems: Noniterative triples for noncanonical
orbitals.
Int. J. Quantum Chem., 55:187–203, 1995.
- 18
-
Roland Lindh.
The reduced multiplication scheme of the Rys–Gauss quadrature
for 1st order integral derivatives.
Theor. Chim. Acta, 85:423–440, 1993.
- 19
-
Shervin Fatehi and Joseph E. Subotnik.
Derivative couplings with built-in electron-translation factors:
Application to benzene.
J. Phys. Chem. Lett., 3(15):2039–2043, 2012.
- 20
-
Michael Stenrup, Roland Lindh, and Ignacio Fdez. Galván.
Constrained numerical gradients and composite gradients: Practical
tools for geometry optimization.
J. Comput. Chem., 36(22):1698–1708, 2015.
- 21
-
Kerstin Andersson, Per-Åke Malmqvist, Björn O. Roos, Andrzej Sadlej,
and Krzysztof Wolinski.
Second-order perturbation theory with a CASSCF reference function.
J. Phys. Chem., 94:5483–5486, 1990.
- 22
-
Kerstin Andersson, Per-Åke Malmqvist, and Björn O. Roos.
Second-order perturbation theory with a complete active space
self-consistent field reference function.
J. Chem. Phys., 96:1218–1226, 1992.
- 23
-
Per Åke Malmqvist, Kristine Pierloot, Abdul Rehaman Moughal Shahi,
Christopher J. Cramer, and Laura Gagliardi.
The restricted active space followed by second-order perturbation
theory method: Theory and application to the study of 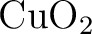 and and
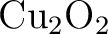 systems. systems.
J. Chem. Phys., 128:204109, 2008.
- 24
-
Vicenta Sauri, Luis Serrano-Andrés, Abdul Rehaman Moughal Shahi, Laura
Gagliardi, Steven Vancoillie, and Kristine Pierloot.
Multiconfigurational second-order perturbation theory restricted
active space (RASPT2) method for electronic excited states: A benchmark
study.
J. Chem. Theory Comput., 7:153–168, 2011.
- 25
-
Björn O. Roos, Markus P. Fülscher, Per-Åke Malmqvist, Manuela
Merchán, and Luis Serrano-Andrés.
Theoretical studies of the electronic spectra of organic molecules.
In Stephen R. Langhoff, editor, Quantum Mechanical Electronic
Structure Calculations with Chemical Accuracy, volume 13 of Understanding Chemical Reactivity, pages 357–438. Kluwer Academic
Publishers, Dordrecht, Netherlands, 1995.
- 26
-
Björn O. Roos, Kerstin Andersson, Markus P. Fülscher, Per-Åke
Malmqvist, Luis Serrano-Andrés, Kristine Pierloot, and Manuela
Merchán.
Multiconfigurational perturbation theory: Applications in
electronic spectroscopy.
In I. Prigogine and Stuart A. Rice, editors, New Methods in
Computational Quantum Mechanics, volume 93 of Advances in Chemical
Physics, pages 213–331. John Wiley & Sons, Hoboken, NJ, USA, 1996.
- 27
-
Kerstin Andersson and Björn O. Roos.
Multiconfigurational second-order perturbation theory: A test of
geometries and binding energies.
Int. J. Quantum Chem., 45:591–607, 1993.
- 28
-
Giovanni Ghigo, Björn O. Roos, and Per-Åke Malmqvist.
A modified definition of the zeroth-order Hamiltonian in
multiconfigurational perturbation theory (CASPT2).
Chem. Phys. Letters, 396:142–149, 2004.
- 29
-
K. Andersson.
Different forms of the zeroth-order Hamiltonian in second-order
perturbation theory with a complete active space self-consistent field
reference function.
Theor. Chim. Acta, 91:31–46, 1995.
- 30
-
Björn O. Roos and Kerstin Andersson.
Multiconfigurational perturbation theory with level shift — the
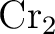 potential revisited. potential revisited.
Chem. Phys. Letters, 245:215–223, 1995.
- 31
-
Björn O. Roos, Kerstin Andersson, Markus P. Fülscher, Luis
Serrano-Andrés, Kristine Pierloot, Manuela Merchán, and Vicent
Molina.
Applications of level shift corrected perturbation theory in
electronic spectroscopy.
J. Mol. Struct. Theochem, 388:257–276, 1996.
- 32
-
Niclas Forsberg and Per-Åke Malmqvist.
Multiconfiguration perturbation theory with imaginary level shift.
Chem. Phys. Letters, 274:196–204, 1997.
- 33
-
Thorstein Thorsteinsson, David L. Cooper, Joseph Gerratt, Peter B. Karadakov,
and Mario Raimondi.
Modern valence bond representations of CASSCF wavefunctions.
Theor. Chim. Acta, 93:343–366, 1996.
- 34
-
David L. Cooper, Thorstein Thorsteinsson, and Joseph Gerratt.
Fully variational optimization of modern VB wave functions using
the CASVB strategy.
Int. J. Quantum Chem., 65:439–451, 1997.
- 35
-
David L. Cooper, Thorstein Thorsteinsson, and Joseph Gerratt.
Modern VB representations of CASSCF wave functions and the
fully-variational optimization of modern VB wave functions using the
CASVB strategy.
Adv. Quantum Chem., 32:51–67, 1998.
- 36
-
T. Thorsteinsson and D. L. Cooper.
An overview of the CASVB approach to modern valence bond
calculations.
In Alfonso Hernández-Laguna, Jean Maruani, Roy McWeeny, and
Stephen Wilson, editors, Quantum Systems in Chemistry and Physics.
Vol. 1: Basic problems and models systems, pages 303–326. Kluwer
Academic Publishers, Dordrecht, Netherlands, 2000.
- 37
-
Pavel Neogrády, Miroslav Urban, and Ivan Hubač.
Spin adapted restricted Hartree–Fock reference coupled cluster
theory for open shell systems.
J. Chem. Phys., 100:3706–3716, 1994.
- 38
-
Pavel Neogrády, Miroslav Urban, and Ivan Hubač.
Spin adapted restricted open shell coupled cluster theory. Linear
version.
J. Chem. Phys., 97:5074–5080, 1992.
- 39
-
Peter J. Knowles, Claudia Hampel, and Hans-Joachim Werner.
Coupled cluster theory for high spin, open shell reference wave
functions.
J. Chem. Phys., 99:5219–5227, 1993.
- 40
-
Miroslav Urban, Jozef Noga, Samuel J. Cole, and Rodney J. Bartlett.
Towards a full CCSDT model for electron correlation.
J. Chem. Phys., 83:4041–4046, 1985.
- 41
-
Krishnan Raghavachari, Gary W. Trucks, John A. Pople, and Martin Head-Gordon.
A fifth-order perturbation comparison of electron correlation
theories.
Chem. Phys. Letters, 157:479–483, 1989.
- 42
-
Reinhart Ahlrichs, Peter Scharf, and Claus Ehrhardt.
The coupled pair functional (CPF). A size consistent modification
of the CI(SD) based on an energy functional.
J. Chem. Phys., 82:890–898, 1985.
- 43
-
Delano P. Chong and Stephen R. Langhoff.
A modified coupled pair functional approach.
J. Chem. Phys., 84:5606–5610, 1986.
- 44
-
Robert J. Gdanitz and Reinhart Ahlrichs.
The averaged coupled-pair functional (ACPF): A size-extensive
modification of MR CI(SD).
Chem. Phys. Letters, 143:413–420, 1988.
- 45
-
B. Roos.
A new method for large-scale CI calculations.
Chem. Phys. Letters, 15:153–159, 1972.
- 46
-
Isaiah Shavitt.
Graph theoretical concepts for the unitary group approach to the
many-electron correlation problem.
Int. J. Quantum Chem., 12-S11:131–148, 1977.
- 47
-
Per E. M. Siegbahn.
Generalizations of the direct CI method based on the graphical
unitary group approach. II. Single and double replacements from any set
of reference configurations.
J. Chem. Phys., 72:1647–1656, 1980.
- 48
-
William C. Swope, Hans C. Andersen, Peter H. Berens, and Kent R. Wilson.
A computer simulation method for the calculation of equilibrium
constants for the formation of physical clusters of molecules: Application
to small water clusters.
J. Chem. Phys., 76:637–649, 1982.
- 49
-
I. V. Abarenkov.
Unit cell for a lattice electrostatic potential.
Phys. Rev. B, 76:165127, 2007.
- 50
-
Peter V. Sushko and Igor V. Abarenkov.
General purpose electrostatic embedding potential.
J. Chem. Theory Comput., 6:1323–1333, 2010.
- 51
-
Jorge M. del Campo and Andreas M. Köster.
A hierarchical transition state search algorithm.
J. Chem. Phys., 129:024107, 2008.
- 52
-
Richard C. Raffenetti.
General contraction of Gaussian atomic orbitals: Core, valence,
polarization, and diffuse basis sets; molecular integral evaluation.
J. Chem. Phys., 58:4452–4458, 1973.
- 53
-
Jan Almlöf and Peter R. Taylor.
General contraction of Gaussian basis sets. I. Atomic natural
orbitals for first- and second-row atoms.
J. Chem. Phys., 86:4070–4077, 1987.
- 54
-
Per-Olof Widmark, Per-Åke Malmqvist, and Björn O. Roos.
Density matrix averaged atomic natural orbital (ANO) basis sets for
correlated molecular wave functions. I. First row atoms.
Theor. Chim. Acta, 77:291, 1990.
- 55
-
Per-Olof Widmark, B. Joakim Persson, and Björn O. Roos.
Density matrix averaged atomic natural orbital (ANO) basis sets for
correlated molecular wave functions. II. Second row atoms.
Theor. Chim. Acta, 79:419–432, 1991.
- 56
-
Rosendo Pou-Amérigo, Manuela Merchán, Ignacio Nebot-Gil, Per-Olof
Widmark, and Björn O. Roos.
Density matrix averaged atomic natural orbital (ANO) basis sets for
correlated molecular wave functions. III. First row transition metal
atoms.
Theor. Chim. Acta, 92:149–181, 1995.
- 57
-
Kristine Pierloot, Birgit Dumez, Per-Olof Widmark, and Björn O. Roos.
Density matrix averaged atomic natural orbital (ANO) basis sets for
correlated molecular wave functions. IV. Medium size basis sets for the
atoms  – –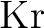 . .
Theor. Chim. Acta, 90:87–114, 1995.
- 58
-
Victor P. Vysotskiy, Jonas Boström, and Valera Veryazov.
A new module for constrained multi-fragment geometry optimization in
internal coordinates implemented in the MOLCAS package.
J. Comput. Chem., 34:2657–2665, 2013.
- 59
-
Yubin Wang, Gaohong Zhai, Binbin Suo, Zhengting Gan, and Zhenyi Wen.
Hole–particle correspondence in CI calculations.
Chem. Phys. Letters, 375:134–140, 2003.
- 60
-
Bing Suo, Gaohong Zhai, Yubin Wang, Zhenyi Wen, Xiangqian Hu, and Lemin Li.
Parallelization of MRCI based on hole–particle symmetry.
J. Comput. Chem., 26:88–96, 2005.
- 61
-
János Pipek and Paul G. Mezey.
A fast intrinsic localization procedure applicable for ab
initio and semiempirical linear combination of atomic orbital wave
functions.
J. Chem. Phys., 90:4916–4926, 1989.
- 62
-
S. F. Boys.
Construction of some molecular orbitals to be approximately invariant
for changes from one molecule to another.
Rev. Mod. Phys., 32:296–299, 1960.
- 63
-
J. M. Foster and S. F. Boys.
Canonical configurational interaction procedure.
Rev. Mod. Phys., 32:300–302, 1960.
- 64
-
Clyde Edmiston and Klaus Ruedenberg.
Localized atomic and molecular orbitals.
Rev. Mod. Phys., 35:457–465, 1963.
- 65
-
Francesco Aquilante, Thomas Bondo Pedersen, Alfredo Sánchez de Merás,
and Henrik Koch.
Fast noniterative orbital localization for large molecules.
J. Chem. Phys., 125:174101, 2006.
- 66
-
Joseph E. Subotnik, Yihan Shao, WanZhen Liang, and Martin Head-Gordon.
An efficient method for calculating maxima of homogeneous functions
of orthogonal matrices: Applications to localized occupied orbitals.
J. Chem. Phys., 121:9220–9229, 2004.
- 67
-
Laura Gagliardi, Roland Lindh, and Gunnar Karlström.
Local properties of quantum chemical systems: The LoProp
approach.
J. Chem. Phys., 121:4494–4500, 2004.
- 68
-
Axel D. Becke and Erin R. Johnson.
Exchange-hole dipole moment and the dispersion interaction.
J. Chem. Phys., 122:154104, 2005.
- 69
-
Anders Bernhardsson, Roland Lindh, Jeppe Olsen, and Markus Fülscher.
A direct implementation of the second-order derivatives of
multiconfigurational SCF energies and an analysis of the preconditioning in
the associated response equation.
Mol. Phys., 96:617–628, 1999.
- 70
-
Jonna Stålring, Anders Bernhardsson, and Roland Lindh.
Analytical gradients of a state average MCSCF state and a state
average diagnostic.
Mol. Phys., 99:103–114, 2001.
- 71
-
Jeppe Olsen, Björn O. Roos, Poul Jørgensen, and Hans Jørgen Aa.
Jensen.
Determinant based configuration interaction algorithms for complete
and restricted configuration interaction spaces.
J. Chem. Phys., 89:2185–2192, 1988.
- 72
-
Giovanni Li Manni, Rebecca K. Carlson, Sijie Luo, Dongxia Ma, Jeppe Olsen,
Donald G. Truhlar, and Laura Gagliardi.
Multi-configuration pair-density functional theory.
J. Chem. Theory Comput., 10:3669–3680, 2014.
- 73
-
Rebecca K. Carlson, Giovanni Li Manni, Andrew L. Sonnenberger, Donald G.
Truhlar, and Laura Gagliardi.
Multiconfiguration pair-density functional theory: Barrier heights
and main group and transition metal energetics.
J. Chem. Theory Comput., 11:82–90, 2015.
- 74
-
Rebecca K. Carlson, Donald G. Truhlar, and Laura Gagliardi.
Multiconfiguration pair-density functional theory: A fully
translated gradient approximation and its performance for transition metal
dimers and the spectroscopy of
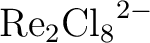 . .
J. Chem. Theory Comput., 11(9):4077, 2015.
- 75
-
Philip W. Anderson.
New approach to the theory of superexchange interactions.
Phys. Rev., 115(1):2–13, Jul 1959.
- 76
-
Philip W. Anderson.
Theory of magnetic exchange interactions: Exchange in insulators
and semiconductors.
In Frederick Seitz and David Turnbull, editors, Solid State
Physics, volume 14, pages 99–214. Academic Press, 1963.
- 77
-
M. E. Lines.
Orbital angular momentum in the theory of paramagnetic clusters.
J. Chem. Phys., 55(6):2977–2984, 1971.
- 78
-
A. Wallqvist, P. Ahlström, and G. Karlström.
New intermolecular energy calculation scheme: Applications to
potential surface and liquid properties of water.
J. Phys. Chem., 94:1649–1656, 1990.
- 79
-
Nigel W. Moriarty and Gunnar Karlström.
Electronic polarization of a water molecule in water. A combined
quantum chemical and statistical mechanical treatment.
J. Phys. Chem., 100:17791–17796, 1996.
- 80
-
Anders Öhrn and Gunnar Karlström.
A theoretical study of the solvent shift to the 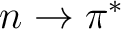 transition in formaldehyde with an effective discrete quantum chemical
solvent model including non-electrostatic perturbation.
transition in formaldehyde with an effective discrete quantum chemical
solvent model including non-electrostatic perturbation.
Mol. Phys., 104:3087–3099, 2006.
- 81
-
Anders Öhrn and Francesco Aquilante.
p-benzoquinone in aqueous solution: Stark shifts in spectra,
asymmetry in solvent structure.
Phys. Chem. Chem. Phys., 9:470–480, 2007.
- 82
-
Anders Öhrn.
Development and Application of a First Principle Molecular Model
for Solvent Effects.
PhD thesis, Lunds Universitet, Theor. Chemistry, Chem. Center, P.O.B.
124,S-221 00 Lund, Sweden, 2008.
- 83
-
Per-Åke Malmqvist, Alistair Rendell, and Björn O. Roos.
The restricted active space self-consistent-field method, implemented
with a split graph unitary group approach.
J. Phys. Chem., 94:5477–5482, 1990.
- 84
-
Björn O. Roos, Peter R. Taylor, and Per E. M. Siegbahn.
A complete active space SCF method (CASSCF) using a density
matrix formulated super-CI approach.
Chem. Phys., 48:157–173, 1980.
- 85
-
Björn O. Roos.
The complete active space self-consistent field method and its
applications in electronic structure calculations.
In K. P. Lawley, editor, Ab Initio Methods in Quantum Chemistry
Part II, volume 69 of Advances in Chemical Physics, pages 399–445.
John Wiley & Sons, Hoboken, NJ, USA, 1987.
- 86
-
Björn O. Roos.
The complete active space SCF method in a Fock-matrix-based
super-CI formulation.
Int. J. Quantum Chem., 18-S14:175–189, 1980.
- 87
-
Francesco Aquilante, Thomas Bondo Pedersen, and Roland Lindh.
Low-cost evaluation of the exchange Fock matrix from Cholesky and
density fitting representations of the electron repulsion integrals.
J. Chem. Phys., 126:194106, 2007.
- 88
-
Per-Åke Malmqvist.
Calculation of transition density matrices by nonunitary orbital
transformations.
Int. J. Quantum Chem., 30:479–494, 1986.
- 89
-
Per-Åke Malmqvist and Björn O. Roos.
The CASSCF state interaction method.
Chem. Phys. Letters, 155:189–194, 1989.
- 90
-
Steven Vancoillie, Per-Åke Malmqvist, and Kristine Pierloot.
Calculation of EPR g tensors for transition-metal complexes based
on multiconfigurational perturbation theory (CASPT2).
ChemPhysChem, 8:1803–1815, 2007.
- 91
-
Steven Vancoillie, Lubomír Rulíšek, Frank Neese, and Kristine
Pierloot.
Theoretical description of the structure and magnetic properties of
nitroxide–Cu(II)–nitroxide spin triads by means of multiconfigurational
ab initio calculations.
J. Phys. Chem. A, 113:6149–6157, 2009.
- 92
-
Chad E. Hoyer, Xuefei Xu, Dongxia Ma, Laura Gagliardi, and Donald G. Truhlar.
Diabatization based on the dipole and quadrupole: The DQ method.
J. Chem. Phys., 141(11):114104, 2014.
- 93
-
Joseph E. Subotnik, Sina Yeganeh, Robert J. Cave, and Mark A. Ratner.
Constructing diabatic states from adiabatic states: extending
generalized Mulliken–Hush to multiple charge centers with Boys
localization.
J. Chem. Phys., 129(24):244101, 2008.
- 94
-
J. Almlöf, K. Faegri, Jr., and K. Korsell.
Principles for a direct SCF approach to LICAO–MO
ab-initio calculations.
J. Comput. Chem., 3:385–399, 1982.
- 95
-
Dieter Cremer and Jürgen Gauss.
An unconventional SCF method for calculations on large molecules.
J. Comput. Chem., 7:274–282, 1986.
- 96
-
Marco Häser and Reinhart Ahlrichs.
Improvements on the direct SCF method.
J. Comput. Chem., 10:104–111, 1989.
- 97
-
Gunnar Karlström.
Dynamical damping based on energy minimization for use ab initio
molecular orbital SCF calculations.
Chem. Phys. Letters, 67:348–350, 1979.
- 98
-
Harrell Sellers.
The C2-DIIS convergence acceleration algorithm.
Int. J. Quantum Chem., 45:31–41, 1993.
- 99
-
Thomas H. Fischer and Jan Almlöf.
General methods for geometry and wave function optimization.
J. Phys. Chem., 96:9768–9774, 1992.
- 100
-
S. H. Vosko, L. Wilk, and M. Nusair.
Accurate spin-dependent electron liquid correlation energies for
local spin density calculations: A critical analysis.
Can. J. Phys., 58:1200–1211, 1980.
- 101
-
A. D. Becke.
Density-functional exchange-energy approximation with correct
asymptotic behavior.
Phys. Rev. A, 38:3098–3100, 1988.
- 102
-
P. Hohenberg and W. Kohn.
Inhomogeneous electron gas.
Phys. Rev., 136:B864–B871, 1964.
- 103
-
W. Kohn and L. J. Sham.
Self-consistent equations including exchange and correlation effects.
Phys. Rev., 140:A1133–A1138, 1965.
- 104
-
J. C. Slater.
Quantum Theory of Molecular and Solids. Vol. 4. The
Self-Consistent Field for Molecular and Solids.
McGraw–Hill, New York, NY, USA, 1974.
- 105
-
A. D. Becke.
Density functional calculations of molecular bond energies.
J. Chem. Phys., 84:4524–4529, 1986.
- 106
-
Axel D. Becke and Erin R. Johnson.
A unified density-functional treatment of dynamical, nondynamical,
and dispersion correlations.
J. Chem. Phys., 127:124108, 2007.
- 107
-
Nicholas C. Handy and Aron J. Cohen.
Left–right correlation energy.
Mol. Phys., 99:403–412, 2001.
- 108
-
Chengteh Lee, Weitao Yang, and Robert G. Parr.
Development of the Colle–Salvetti correlation-energy formula
into a functional of the electron density.
Phys. Rev. B, 37:785–789, 1988.
- 109
-
Burkhard Miehlich, Andreas Savin, Hermann Stoll, and Heinzwerner Preuss.
Results obtained with the correlation energy density functionals of
Becke and Lee, Yang and Parr.
Chem. Phys. Letters, 157:200–206, 1989.
- 110
-
John P. Perdew, Kieron Burke, and Matthias Ernzerhof.
Generalized gradient approximation made simple.
Phys. Rev. Letters, 77:3865–3868, 1996.
- 111
-
Axel D. Becke.
Density-functional thermochemistry. III. The role of exact
exchange.
J. Chem. Phys., 98:5648–5652, 1993.
- 112
-
Stefan Grimme.
Semiempirical hybrid density functional with perturbative
second-order correlation.
J. Chem. Phys., 124:034108, 2006.
- 113
-
Phillip A. Stewart and Peter M. W. Gill.
Becke–Wigner: A simple but powerful density functional.
J. Chem. Soc. Faraday Trans., 91:4337–4341, 1995.
- 114
-
Peter M. W. Gill.
A new gradient-corrected exchange functional.
Mol. Phys., 89:433–445, 1996.
- 115
-
Wee-Meng Hoe, Aaron J. Cohen, and Nicholas C. Handy.
Assessment of a new local exchange functional OPTX.
Chem. Phys. Letters, 341:319–328, 2001.
- 116
-
Mark J. Allen, Thomas W. Keal, and David J. Tozer.
Improved NMR chemical shifts in density functional theory.
Chem. Phys. Letters, 380:70–77, 2003.
- 117
-
Thomas W. Keal and David J. Tozer.
A semiempirical generalized gradient approximation
exchange-correlation functional.
J. Chem. Phys., 121:5654–5660, 2004.
- 118
-
John P. Perdew, Matthias Ernzerhof, and Kieron Burke.
Rationale for mixing exact exchange with density functional
approximations.
J. Chem. Phys., 105:9982–9985, 1996.
- 119
-
Adrienn Ruzsinszky, Gábor I. Csonka, and Gustavo E. Scuseria.
Regularized gradient expansion for atoms, molecules, and solids.
J. Chem. Theory Comput., 5:763–769, 2009.
- 120
-
Vincent Tognetti, Pietro Cortona, and Carlo Adamo.
A new parameter-free correlation functional based on an average
atomic reduced density gradient analysis.
J. Chem. Phys., 128:034101, 2008.
- 121
-
Marcel Swart, Miquel Solà, and F. Matthias Bickelhaupt.
A new all-round density functional based on spin states and
SN2 barriers.
J. Chem. Phys., 131:049103, 2009.
- 122
-
Yan Zhao and Donald G. Truhlar.
A new local density functional for main-group thermochemistry,
transition metal bonding, thermochemical kinetics, and noncovalent
interactions.
J. Chem. Phys., 125:194101, 2006.
- 123
-
Yan Zhao and Donald G. Truhlar.
Density functional for spectroscopy: No long-range self-interaction
error, good performance for Rydberg and charge-transfer states, and better
performance on average than B3LYP for ground states.
J. Phys. Chem. A, 110:13126–13130, 2006.
- 124
-
Yan Zhao and Donald G. Truhlar.
The M06 suite of density functionals for main group
thermochemistry, thermochemical kinetics, noncovalent interactions, excited
states, and transition elements: Two new functionals and systematic testing
of four M06-class functionals and 12 other functionals.
Theor. Chem. Acc., 120:215–241, 2008.
- 125
-
Yan Zhao and Donald G. Truhlar.
Density functionals with broad applicability in chemistry.
Acc. Chem. Res., 41:157–167, 2008.
- 126
-
R. Lindh, U. Ryu, and B. Liu.
The reduced multiplication scheme of the Rys quadrature and new
recurrence relations for auxiliary function based two-electron integral
evaluation.
J. Chem. Phys., 95:5889–5897, 1991.
- 127
-
Ernest R. Davidson.
Use of double cosets in constructing integrals over symmetry
orbitals.
J. Chem. Phys., 62:400–403, 1975.
- 128
-
Benny G. Johnson, Peter M. W. Gill, and John A. Pople.
The performance of a family of density functional methods.
J. Chem. Phys., 98:5612–5626, 1993.
- 129
-
Nicholas C. Handy, David J. Tozer, Gregory J. Laming, Christopher W. Murray,
and Roger D. Amos.
Analytic second derivatives of the potential energy surface.
Isr. J. Chem., 33:331–344, 1993.
- 130
-
Jon Baker, Jan Andzelm, Andrew Scheiner, and Bernard Delley.
The effect of grid quality and weight derivatives in density
functional calculations.
J. Chem. Phys., 101:8894–8902, 1994.
- 131
-
Michael E. Mura and Peter J. Knowles.
Improved radial grids for quadrature in molecular density-functional
calculations.
J. Chem. Phys., 104:9848–9858, 1996.
- 132
-
A. D. Becke.
A multicenter numerical integration scheme for polyatomic molecules.
J. Chem. Phys., 88:2547–2553, 1988.
- 133
-
Christopher W. Murray, Nicholas C. Handy, and Gregory J. Laming.
Quadrature schemes for integrals of density functional theory.
Mol. Phys., 78:997–1014, 1993.
- 134
-
Oliver Treutler and Reinhart Ahlrichs.
Efficient molecular numerical integration schemes.
J. Chem. Phys., 102:346–354, 1995.
- 135
-
Roland Lindh, Per-Åke Malmqvist, and Laura Gagliardi.
Molecular integrals by numerical quadrature. I. Radial
integration.
Theor. Chem. Acc., 106:178–187, 2001.
- 136
-
Daoling Peng and Markus Reiher.
Exact decoupling of the relativistic Fock operator.
Theor. Chem. Acc., 131:1081, 2012.
- 137
-
Daoling Peng and Kimihiko Hirao.
An arbitrary order Douglas–Kroll method with polynomial cost.
J. Chem. Phys., 130:044102, 2009.
- 138
-
Markus Reiher and Alexander Wolf.
Exact decoupling of the Dirac Hamiltonian. I. General theory.
J. Chem. Phys., 121:2037–2047, 2004.
- 139
-
Markus Reiher and Alexander Wolf.
Exact decoupling of the Dirac Hamiltonian. II. The
generalized Douglas–Kroll–Hess transformation up to arbitrary order.
J. Chem. Phys., 121:10945–10956, 2004.
- 140
-
Markus Reiher.
Douglas–Kroll–Hess theory: a relativistic electrons-only
theory for chemistry.
Theor. Chem. Acc., 116:241–252, 2006.
- 141
-
Alexander Wolf, Markus Reiher, and Bernd Artur Hess.
The generalized Douglas–Kroll transformation.
J. Chem. Phys., 117:9215–9226, 2002.
- 142
-
Alexander Wolf and Markus Reiher.
Exact decoupling of the Dirac Hamiltonian. III. Molecular
properties.
J. Chem. Phys., 124:064102, 2006.
- 143
-
Alexander Wolf and Markus Reiher.
Exact decoupling of the Dirac Hamiltonian. IV. Automated
evaluation of molecular properties within the Douglas–Kroll–Hess
theory up to arbitrary order.
J. Chem. Phys., 124:064103, 2006.
- 144
-
Werner Kutzelnigg and Wenjian Liu.
Quasirelativistic theory equivalent to fully relativistic theory.
J. Chem. Phys., 123:241102, 2005.
- 145
-
Wenjian Liu and Daoling Peng.
Infinite-order quasirelativistic density functional method based on
the exact matrix quasirelativistic theory.
J. Chem. Phys., 125:044102, 2006.
- 146
-
Daoling Peng, Wenjian Liu, Yunlong Xiao, and Lan Cheng.
Making four- and two-component relativistic density functional
methods fully equivalent based on the idea of "from atoms to molecule".
J. Chem. Phys., 127:104106, 2007.
- 147
-
Maria Barysz, Andrzej J. Sadlej, and Jaap G. Snijders.
Nonsingular two/one-component relativistic Hamiltonians accurate
through arbitrary high order in 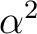 . .
Int. J. Quantum Chem., 65:225–239, 1997.
- 148
-
Dariusz Kędziera and Maria Barysz.
Non-iterative approach to the infinite-order two-component (IOTC)
relativistic theory and the non-symmetric algebraic Riccati equation.
Chem. Phys. Letters, 446:176–181, 2007.
- 149
-
Daoling Peng and Markus Reiher.
Local relativistic exact decoupling.
J. Chem. Phys., 136:244108, 2012.
- 150
-
Liviu F. Chibotaru, Liviu Ungur, and Alessandro Soncini.
The origin of nonmagnetic Kramers doublets in the ground state of
dysprosium triangles: Evidence for a toroidal magnetic moment.
Angew. Chem. Int. Ed., 47:4126–4129, 2008.
- 151
-
Liviu F. Chibotaru, Liviu Ungur, Christophe Aronica, Hani Elmoll, Guillaume
Pillet, and Dominique Luneau.
Structure, magnetism, and theoretical study of a mixed-valence
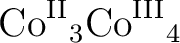 heptanuclear wheel: Lack of SMM behavior
despite negative magnetic anisotropy. heptanuclear wheel: Lack of SMM behavior
despite negative magnetic anisotropy.
J. Am. Chem. Soc., 130:12445–12455, 2008.
- 152
-
Liviu F. Chibotaru and Liviu Ungur.
Ab initio calculation of anisotropic magnetic properties of
complexes. I. Unique definition of pseudospin Hamiltonians and their
derivation.
J. Chem. Phys., 137:064112, 2012.
- 153
-
Liviu Ungur and Liviu F. Chibotaru.
Ab initio crystal field for lanthanides.
Chem. Eur. J., 23(15):3708–3718, 2017.
- 154
-
Czeslaw Rudowicz.
Transformation relations for the conventional okq and normalised
o'kq Stevens Operator Equivalents with k=1 to 6 and
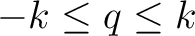 . .
J. Phys. C: Solid State Phys., 18(7):1415, 1985.
- 155
-
C. Rudowicz and C. Y. Chung.
Generalization of the extended Stevens operators to higher ranks
and spins and systematic review of the tables of the tensor operators and
their matrix elements.
J. Phys.: Condens. Matter, 16(32):5825, 2004.
- 156
-
Czeslaw Rudowicz and Miroslav Karbowiak.
Disentangling intricate web of interrelated notions at the interface
between the physical (crystal field) Hamiltonians and the
effective (spin) Hamiltonians.
Coord. Chem. Rev., 287:28, 2015.
- 157
-
Roland Lindh, Anders Bernhardsson, Gunnar Karlström, and Per-Åke
Malmqvist.
On the use of a Hessian model function in molecular geometry
optimizations.
Chem. Phys. Letters, 241:423–428, 1995.
- 158
-
Chunyang Peng, Philippe Y. Ayala, H. Bernhard Schlegel, and Michael J. Frisch.
Using redundant internal coordinates to optimize equilibrium
geometries and transition states.
J. Comput. Chem., 17:49–56, 1996.
- 159
-
P. Pulay and G. Fogarasi.
Geometry optimization in redundant internal coordinates.
J. Chem. Phys., 96:2856–2860, 1992.
- 160
-
Jon Baker, Alain Kessi, and Bernard Delley.
The generation and use of delocalized internal coordinates in
geometry optimization.
J. Chem. Phys., 105:192–212, 1996.
- 161
-
Roland Lindh, Anders Bernhardsson, and Martin Schütz.
Force-constant weighted redundant coordinates in molecular geometry
optimizations.
Chem. Phys. Letters, 303:567–575, 1999.
- 162
-
Jon Baker.
Techniques for geometry optimization: A comparison of Cartesian
and natural internal coordinates.
J. Comput. Chem., 14:1085–1100, 1993.
- 163
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A.
Robb, J. R. Cheeseman, T. A. Keith, G. A. Petersson, J. A. Montgomery, Jr.,
K. Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B.
Foresman, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe,
C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle,
R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker,
J. P. Stewart, M. Head-Gordon, C. Gonzalez, and J. A. Pople.
Gaussian 94 (Revision A.1).
Gaussian, Inc., Pittsburgh, PA, USA, 1995.
- 164
-
Josep Maria Bofill.
Updated Hessian matrix and the restricted step method for locating
transition structures.
J. Comput. Chem., 15:1–11, 1994.
- 165
-
Josep Maria Bofill.
Remarks on the updated Hessian matrix methods.
Int. J. Quantum Chem., 94:324–332, 2003.
- 166
-
Ajit Banerjee, Noah Adams, Jack Simons, and Ron Shepard.
Search for stationary points on surfaces.
J. Phys. Chem., 89:52–57, 1985.
- 167
-
Emili Besalú and Josep Maria Bofill.
On the automatic restricted-step rational-function-optimization
method.
Theor. Chem. Acc., 100:265–274, 1998.
- 168
-
Pál Császár and Péter Pulay.
Geometry optimization by direct inversion in the iterative subspace.
J. Mol. Struct., 114:31–34, 1984.
- 169
-
Péter Pulay.
Convergence acceleration of iterative sequences. The case of SCF
iteration.
Chem. Phys. Letters, 73:393–398, 1980.
- 170
-
P. Pulay.
Improved SCF convergence acceleration.
J. Comput. Chem., 3:556–560, 1982.
- 171
-
Charles J. Cerjan and William H. Miller.
On finding transition states.
J. Chem. Phys., 75:2800–2806, 1981.
- 172
-
Satoshi Maeda, Koichi Ohno, and Keiji Morokuma.
Updated branching plane for finding conical intersections without
coupling derivative vectors.
J. Chem. Theory Comput., 6(5):1538–1545, 2010.
- 173
-
John C. Tully.
Molecular dynamics with electronic transitions.
J. Chem. Phys., 93(2):1061–1071, 1990.
- 174
-
Sharon Hammes-Schiffer and John C. Tully.
Proton transfer in solution: Molecular dynamics with quantum
transitions.
J. Chem. Phys., 101(6):4657–4667, 1994.
- 175
-
Giovanni Granucci and Maurizio Persico.
Critical appraisal of the fewest switches algorithm for surface
hopping.
J. Chem. Phys., 126(13):134114, 2007.
- 176
-
F. Plasser, M. Wormit, S. A. Mewes, B. Thomitzni, and A. Dreuw.
libwfa: Wave-function analysis tool library for quantum chemical
applications.
- 177
-
Felix Plasser, Stefanie A. Mewes, Andreas Dreuw, and Leticia González.
Detailed wave function analysis for multireference methods:
Implementation in the Molcas program package and applications to
tetracene.
J. Chem. Theory Comput., 13(11):5343–5353, 2017.
- 178
-
Richard L. Martin.
Natural transition orbitals.
J. Chem. Phys., 118(11):4775–4777, 2003.
- 179
-
Felix Plasser, Michael Wormit, and Andreas Dreuw.
New tools for the systematic analysis and visualization of electronic
excitations. I. Formalism.
J. Chem. Phys., 141(2):024106, 2014.
- 180
-
Felix Plasser, Stefanie A. Bäppler, Michael Wormit, and Andreas Dreuw.
New tools for the systematic analysis and visualization of electronic
excitations. II. Applications.
J. Chem. Phys., 141(2):024107, 2014.
- 181
-
Stefanie A. Bäppler, Felix Plasser, Michael Wormit, and Andreas Dreuw.
Exciton analysis of many-body wave functions: Bridging the gap
between the quasiparticle and molecular orbital pictures.
Phys. Rev. A, 90(5):052521, 2014.
- 182
-
Felix Plasser, Benjamin Thomitzni, Stefanie A. Bäppler, Jan Wenzel, Dirk R.
Rehn, Michael Wormit, and Andreas Dreuw.
Statistical analysis of electronic excitation processes: Spatial
location, compactness, charge transfer, and electron-hole correlation.
J. Comput. Chem., 36(21):1609–1620, 2015.
- 183
-
Felix Plasser and Hans Lischka.
Analysis of excitonic and charge transfer interactions from quantum
chemical calculations.
J. Chem. Theory Comput., 8(8):2777–2789, 2012.
- 184
-
F. Plasser.
TheoDORE: a package for theoretical density, orbital relaxation,
and exciton analysis.
- 185
-
Martin Head-Gordon.
Characterizing unpaired electrons from the one-particle density
matrix.
Chem. Phys. Letters, 372(3-4):508–511, 2003.
- 186
-
Felix Plasser.
Entanglement entropy of electronic excitations.
J. Chem. Phys., 144(19):194107, 2016.
- 187
-
Andrzej J. Sadlej.
Medium-size polarized basis sets for high-level correlated
calculations of molecular electric properties.
Collect. Czech. Chem. Commun., 53:1995–2016, 1988.
- 188
-
Andrzej J. Sadlej.
Medium-size polarized basis sets for high-level-correlated
calculations of molecular electric properties. II. Second-row atoms:
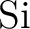 through through 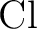 . .
Theor. Chim. Acta, 79:123–140, 1991.
- 189
-
Andrzej J. Sadlej and Miroslav Urban.
Medium-size polarized basis sets for high-level-correlated
calculations of molecular electric properties: III. Alkali (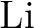 , ,
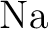 , , 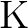 , , 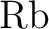 ) and alkaline-earth ( ) and alkaline-earth (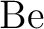 , , 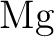 , ,
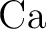 , , 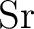 ) atoms. ) atoms.
J. Mol. Struct. Theochem, 234:147–171, 1991.
- 190
-
Andrzej J. Sadlej.
Medium-size polarized basis sets for high-level-correlated
calculations of molecular electric properties. IV. Third-row atoms:
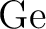 through through 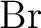 . .
Theor. Chim. Acta, 81:45–63, 1991.
- 191
-
Andrzej J. Sadlej.
Medium-size polarized basis sets for high-level-correlated
calculations of molecular electric properties. V. Fourth-row atoms:
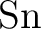 through through 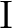 . .
Theor. Chim. Acta, 81:339–354, 1992.
- 192
-
Vladimir Kellö and Andrzej J. Sadlej.
Estimates of relativistic contributions to molecular properties.
J. Chem. Phys., 93:8122–8132, 1990.
- 193
-
Andrzej J. Sadlej and Miroslav Urban.
Mutual dependence of relativistic and electron correlation
contributions to molecular properties: The dipole moment of 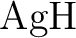 . .
Chem. Phys. Letters, 176:293–302, 1991.
- 194
-
Sigeru Huzinaga, Luis Seijo, Zoila Barandiarán, and Mariusz Klobukowski.
The ab initio model potential method. Main group elements.
J. Chem. Phys., 86:2132–2145, 1987.
- 195
-
Zoila Barandiarán and Luis Seijo.
The ab initio model potential representation of the
crystalline environment. Theoretical study of the local distortion on
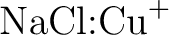 . .
J. Chem. Phys., 89:5739–5746, 1988.
- 196
-
Zoila Barandiarán, Luis Seijo, and Sigeru Huzinaga.
The ab initio model potential method. Second series
transition metal elements.
J. Chem. Phys., 93:5843–5850, 1990.
- 197
-
Christina Wittborn and Ulf Wahlgren.
New relativistic effective core potentials for heavy elements.
Chem. Phys., 201:357–362, 1995.
- 198
-
Frank Rakowitz, Christel M. Marian, Luis Seijo, and Ulf Wahlgren.
Spin-free relativistic no-pair ab initio core model potentials
and valence basis sets for the transition metal elements 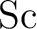 to to
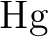 . Part I. . Part I.
J. Chem. Phys., 110:3678–3686, 1999.
- 199
-
Frank Rakowitz, Christel M. Marian, and Luis Seijo.
Spin-free relativistic no-pair ab initio core model potentials
and valence basis sets for the transition metal elements to
. II.
J. Chem. Phys., 111:10436–10443, 1999.
- 200
-
Z. Barandiarán and L. Seijo.
Local properties of imperfect crystals.
In S. Fraga, editor, Computational Chemistry: Structure,
Interactions and Reactivity, volume 77B of Studies in Physical and
Theoretical Chemistry, pages 435–461. Elsevier, Amsterdam, Netherlands,
1992.
- 201
-
James C. Phillips and Leonard Kleinman.
New method for calculating wave functions in crystals and molecules.
Phys. Rev., 116:287–294, 1959.
- 202
-
S. Huzinaga and A. A. Cantu.
Theory of separability of many-electron systems.
J. Chem. Phys., 55:5543–5549, 1971.
- 203
-
Sigeru Huzinaga, Dennis McWilliams, and Antonio A. Cantu.
Projection operators in Hartree–Fock theory.
Adv. Quantum Chem., 7:187–220, 1973.
- 204
-
José Luis Pascual, Luis Seijo, and Zoila Barandiarán.
Ab initio model potential study of environmental effects on the
Jahn–Teller parameters of 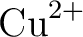 and and 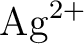 impurities
in impurities
in 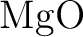 , , 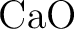 , and , and 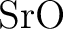 hosts. hosts.
J. Chem. Phys., 98:9715–9724, 1993.
- 205
-
M. Pelissier, N. Komiha, and J.-P. Daudey.
One-center expansion for pseudopotential matrix elements.
J. Comput. Chem., 9:298–302, 1988.
- 206
-
P. Jeffrey Hay and Willard R. Wadt.
Ab initio effective core potentials for molecular
calculations. Potentials for the transition metal atoms to
.
J. Chem. Phys., 82:270–283, 1985.
- 207
-
P. Jeffrey Hay and Willard R. Wadt.
Ab initio effective core potentials for molecular
calculations. Potentials for main group elements to 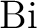 . .
J. Chem. Phys., 82:284–298, 1985.
- 208
-
P. Jeffrey Hay and Willard R. Wadt.
Ab initio effective core potentials for molecular
calculations. Potentials for to 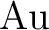 including the outermost
core orbitals. including the outermost
core orbitals.
J. Chem. Phys., 82:299–310, 1985.
- 209
-
Patricio Fuentealba, Heinzwerner Preuss, Hermann Stoll, and László
Von Szentpály.
A proper account of core-polarization with pseudopotentials: Single
valence-electron alkali compounds.
Chem. Phys. Letters, 89:418–422, 1982.
- 210
-
P. Fuentealba, L. von Szentpály, H. Preuss, and H. Stoll.
Pseudopotential calculations for alkaline-earth atoms.
J. Phys. B: At. Mol. Phys., 18:1287–1296, 1985.
- 211
-
G. Igel-Mann, H. Stoll, and H. Preuss.
Pseudopotentials for main group elements (IIIa through VIIa).
Mol. Phys., 65:1321–1328, 1988.
- 212
-
Andreas Bergner, Michael Dolg, Wolfgang Küchle, Hermann Stoll, and
Heinzwerner Preuß.
Ab initio energy-adjusted pseudopotentials for elements of
groups 13–17.
Mol. Phys., 80:1431–1441, 1993.
- 213
-
P. Fuentealba, H. Stoll, L. von Szentpály, P. Schwerdtfeger, and H. Preuss.
On the reliability of semi-empirical pseudopotentials: Simulation
of Hartree–Fock and Dirac–Fock results.
J. Phys. B: At. Mol. Phys., 16:L323–L328, 1983.
- 214
-
M. Kaupp, P. v. R. Schleyer, H. Stoll, and H. Preuss.
Pseudopotential approaches to , , and 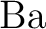 hydrides. Why are some alkaline earth
hydrides. Why are some alkaline earth 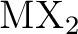 compounds bent? compounds bent?
J. Chem. Phys., 94:1360–1366, 1991.
- 215
-
M. Dolg, U. Wedig, H. Stoll, and H. Preuss.
Energy-adjusted ab initio pseudopotentials for the first row
transition elements.
J. Chem. Phys., 86:866–872, 1987.
- 216
-
Ulrich Wedig, Michael Dolg, Hermann Stoll, and Heinzwerner Preuss.
Energy-adjusted pseudopotentials for transition-metal elements.
In A. Veillard, editor, Quantum Chemistry: The Challenge of
Transition Metals and Coordination Chemistry, volume 176 of NATO ASI
Series, pages 79–89. D. Reidel, Dordrecht, Netherlands, 1986.
- 217
-
László Von Szentpály, Patricio Fuentealba, Heinzwerner Preuss, and
Hermann Stoll.
Pseudopotential calculations on 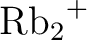 , , 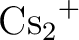 , ,
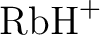 , , 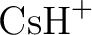 and the mixed alkali dimer ions. and the mixed alkali dimer ions.
Chem. Phys. Letters, 93:555–559, 1982.
- 218
-
D. Andrae, U. Häußermann, M. Dolg, H. Stoll, and H. Preuß.
Energy-adjusted ab initio pseudopotentials for the second and
third row transition elements.
Theor. Chim. Acta, 77:123–141, 1990.
- 219
-
H. Stoll, P. Fuentealba, P. Schwerdtfeger, J. Flad, L. v. Szentpály, and
H. Preuss.
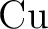 and and 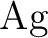 as one-valence-electron atoms: CI results
and quadrupole corrections for as one-valence-electron atoms: CI results
and quadrupole corrections for 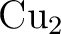 , , 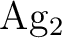 , , 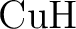 , and
. , and
.
J. Chem. Phys., 81:2732–2736, 1984.
- 220
-
W. Küchle, M. Dolg, H. Stoll, and H. Preuss.
Ab initio pseudopotentials for through 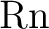 .
I. Parameter sets and atomic calculations. .
I. Parameter sets and atomic calculations.
Mol. Phys., 74:1245–1263, 1991.
- 221
-
Gudrun Igel-Mann.
Semiempirische Pseudopotentiale; Untersuchungen an
Hauptgruppenelementen und Nebengruppenelementen mit abgeschlossener
d-Schale.
PhD thesis, Universität Stuttgart, Institut für Theoretische
Chemie, 1987.
- 222
-
M. Dolg, H. Stoll, and H. Preuss.
A combination of quasirelativistic pseudopotential and ligand field
calculations for lanthanoid compounds.
Theor. Chim. Acta, 85:441–450, 1993.
- 223
-
M. Dolg, H. Stoll, and H. Preuss.
Energy-adjusted ab initio pseudopotentials for the rare earth
elements.
J. Chem. Phys., 90:1730–1734, 1989.
- 224
-
Michael Dolg, Peter Fulde, Wolfgang Küchle, Carl-Stefan Neumann, and
Hermann Stoll.
Ground state calculations of di-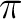 -cyclooctatetraene cerium. -cyclooctatetraene cerium.
J. Chem. Phys., 94:3011–3017, 1991.
- 225
-
M. Dolg, H. Stoll, A. Savin, and H. Preuss.
Energy-adjusted pseudopotentials for the rare earth elements.
Theor. Chim. Acta, 75:173–194, 1989.
- 226
-
Michael Dolg, Hermann Stoll, Heinz-Jürgen Flad, and Heinzwerner Preuss.
Ab initio pseudopotential study of 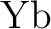 and and 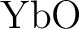 . .
J. Chem. Phys., 97:1162–1173, 1992.
- 227
-
Michael Dolg, Hermann Stoll, Heinzwerner Preuss, and Russell M. Pitzer.
Relativistic and correlation effects for element 105 (hahnium,
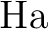 ): A comparative study of ): A comparative study of 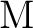 and and 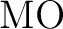 ( = ( =
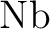 , , 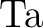 , ) using energy-adjusted ab initio
pseudopotentials. , ) using energy-adjusted ab initio
pseudopotentials.
J. Phys. Chem., 97:5852–5859, 1993.
- 228
-
Peter Schwerdtfeger, Michael Dolg, W. H. Eugen Schwarz, Graham A. Bowmaker, and
Peter D. W. Boyd.
Relativistic effects in gold chemistry. I. Diatomic gold
compounds.
J. Chem. Phys., 91:1762–1774, 1989.
- 229
-
U. Häussermann, M. Dolg, H. Stoll, H. Preuss, P. Schwerdtfeger, and R. M.
Pitzer.
Accuracy of energy-adjusted quasirelativistic ab initio
pseudopotentials. All-electron and pseudopotential benchmark calculations
for , 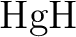 and their cations. and their cations.
Mol. Phys., 78:1211–1224, 1993.
- 230
-
W. Küchle, M. Dolg, H. Stoll, and H. Preuss.
Energy-adjusted pseudopotentials for the actinides. Parameter sets
and test calculations for thorium and thorium monoxide.
J. Chem. Phys., 100:7535–7542, 1994.
- 231
-
Andreas Nicklass, Michael Dolg, Hermann Stoll, and Heinzwerner Preuss.
Ab initio energy-adjusted pseudopotentials for the noble gases
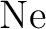 through through 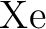 : Calculation of atomic dipole and quadrupole
polarizabilities. : Calculation of atomic dipole and quadrupole
polarizabilities.
J. Chem. Phys., 102:8942–8952, 1995.
- 232
-
Thierry Leininger, Andreas Nicklass, Hermann Stoll, Michael Dolg, and Peter
Schwerdtfeger.
The accuracy of the pseudopotential approximation. II. A
comparison of various core sizes for indium pseudopotentials in calculations
for spectroscopic constants of 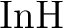 , , 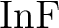 , and , and 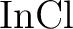 . .
J. Chem. Phys., 105:1052–1059, 1996.
- 233
-
Xiaoyan Cao, Michael Dolg, and Hermann Stoll.
Valence basis sets for relativistic energy-consistent small-core
actinide pseudopotentials.
J. Chem. Phys., 118:487–496, 2003.
- 234
-
L. R. Kahn and W. A. Goddard, III.
A direct test of the validity of the use of pseudopotentials in
molecules.
Chem. Phys. Letters, 2:667–670, 1968.
- 235
-
Phillip A. Christiansen, Yoon S. Lee, and Kenneth S. Pitzer.
Improved ab initio effective core potentials for molecular
calculations.
J. Chem. Phys., 71:4445–4450, 1979.
- 236
-
Philippe Durand and Jean-Claude Barthelat.
A theoretical method to determine atomic pseudopotentials for
electronic structure calculations of molecules and solids.
Theor. Chim. Acta, 38:283–302, 1975.
- 237
-
Chris-Kriton Skylaris, Laura Gagliardi, Nicholas C. Handy, Andrew G. Ioannou,
Steven Spencer, Andrew Willetts, and Adrian M. Simper.
An efficient method for calculating effective core potential
integrals which involve projection operators.
Chem. Phys. Letters, 296:445–451, 1998.
- 238
-
Harry Partridge, Stephen R. Langhoff, and Charles W. Bauschlicher, Jr.
Electronic spectroscopy of diatomic molecules.
In Stephen R. Langhoff, editor, Quantum Mechanical Electronic
Structure Calculations with Chemical Accuracy, volume 13 of Understanding Chemical Reactivity, pages 209–260. Kluwer Academic
Publishers, Dordrecht, Netherlands, 1995.
- 239
-
Peter R. Taylor.
Molecular symmetry and quantum chemistry.
In Björn O. Roos, editor, Lecture Notes in Quantum
Chemistry. European Summer School in Quantum Chemistry, volume 58 of Lecture Notes in Chemistry, pages 89–176. Springer-Verlag, Berlin, Germany,
1992.
- 240
-
Margareta R. A. Blomberg, Per E. M. Siegbahn, and Björn O. Roos.
A theoretical study of 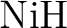 optical spectrum and potential
curves. optical spectrum and potential
curves.
Mol. Phys., 47:127–143, 1982.
- 241
-
Rosendo Pou-Amérigo, Manuela Merchán, Ignacio Nebot-Gil, Per-Åke
Malmqvist, and Björn O. Roos.
The chemical bonds in , , , and
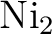 studied with multiconfigurational second order perturbation
theory. studied with multiconfigurational second order perturbation
theory.
J. Chem. Phys., 101:4893–4902, 1994.
- 242
-
Kerstin Andersson and Björn O. Roos.
Excitation energies in the nickel atom studied with the complete
active space SCF method and second-order perturbation theory.
Chem. Phys. Letters, 191:507–514, 1992.
- 243
-
Gerhard Herzberg.
Molecular Spectra and Molecular Structure. Vol I. Spectra
of Diatomic Molecules.
D. Van Nostrand, Princeton, NJ, USA, 2nd edition, 1966.
- 244
-
Peter R. Taylor.
Accurate calculations and calibration.
In Björn O. Roos, editor, Lecture Notes in Quantum
Chemistry. European Summer School in Quantum Chemistry, volume 58 of Lecture Notes in Chemistry, pages 325–412. Springer-Verlag, Berlin,
Germany, 1992.
- 245
-
Remedios González-Luque, Manuela Merchán, and Björn O. Roos.
A theoretical determination of the dissociation energy of the nitric
oxide dimer.
Theor. Chim. Acta, 88:425–435, 1994.
- 246
-
M. Perić, B. Engels, and S. D. Peyerimhoff.
Theoretical spectroscopy on small molecules: Ab initio
investigations of vibronic structure, spin–orbit splittings and magnetic
hyperfine effects in the electronic spectra of triatomic molecules.
In Stephen R. Langhoff, editor, Quantum Mechanical Electronic
Structure Calculations with Chemical Accuracy, volume 13 of Understanding Chemical Reactivity, pages 261–356. Kluwer Academic
Publishers, Dordrecht, Netherlands, 1995.
- 247
-
Trygve Helgaker.
Optimization of minima and saddle points.
In Björn O. Roos, editor, Lecture Notes in Quantum
Chemistry. European Summer School in Quantum Chemistry, volume 58 of Lecture Notes in Chemistry, pages 295–324. Springer-Verlag, Berlin,
Germany, 1992.
- 248
-
Mercedes Rubio, Manuela Merchán, Enrique Ortí, and Björn O. Roos.
A theoretical study of the electronic spectrum of naphthalene.
Chem. Phys., 179:395–409, 1994.
- 249
-
Luis Serrano-Andrés, Manuela Merchán, Ignacio Nebot-Gil, Roland Lindh,
and Björn O. Roos.
Towards an accurate molecular orbital theory for excited states:
Ethene, butadiene, and hexatriene.
J. Chem. Phys., 98:3151–3162, 1993.
- 250
-
Luis Serrano-Andrés and Björn O. Roos.
Theoretical study of the absorption and emission spectra of indole in
the gas phase and in a solvent.
J. Am. Chem. Soc., 118:185–195, 1996.
- 251
-
Christopher S. Page, Manuela Merchán, Luis Serrano-Andrés, and Massimo
Olivucci.
A theoretical study of the low-lying excited states of trans-
and cis-urocanic acid.
J. Phys. Chem. A, 103:9864–9871, 1999.
- 252
-
Rosendo Pou-Amérigo, Manuela Merchán, and Enrique Ortí.
Theoretical study of the electronic spectrum of
p-benzoquinone.
J. Chem. Phys., 110:9536–9546, 1999.
- 253
-
C. E. Blom and A. Bauder.
Microwave spectrum, rotational constants and dipole moment of
s-cis acrolein.
Chem. Phys. Letters, 88:55–58, 1982.
- 254
-
Vicent Molina and Manuela Merchán.
Theoretical analysis of the electronic spectra of benzaldehyde.
J. Phys. Chem. A, 105:3745–3751, 2001.
- 255
-
Francis Ford, Tetsuro Yuzawa, Matthew S. Platz, Stephan Matzinger, and Markus
Fülscher.
Rearrangement of dimethylcarbene to propene: Study by laser flash
photolysis and ab Initio molecular orbital theory.
J. Am. Chem. Soc., 120:4430–4438, 1998.
- 256
-
Timothy J. Lee and Peter R. Taylor.
A diagnostic for determining the quality of single-reference electron
correlation methods.
Int. J. Quantum Chem., 36-S23:199–207, 1989.
- 257
-
Timothy J. Lee and Gustavo E. Scuseria.
Achieving chemical accuracy with coupled-cluster theory.
In Stephen R. Langhoff, editor, Quantum Mechanical Electronic
Structure Calculations with Chemical Accuracy, volume 13 of Understanding Chemical Reactivity, pages 47–108. Kluwer Academic
Publishers, Dordrecht, Netherlands, 1995.
- 258
-
S. Matzinger and M. P. Fülscher.
Methyl substitution in carbenes. A theoretical prediction of the
singlet–triplet energy separation of dimethylcarbene.
J. Phys. Chem., 99:10747–10751, 1995.
- 259
-
David J. Tozer, Roger D. Amos, Nicholas C. Handy, Björn O. Roos, and Luis
Serrano-Andrés.
Does density functional theory contribute to the understanding of
excited states of unsaturated organic compounds?
Mol. Phys., 97:859–868, 1999.
- 260
-
Luis Serrano-Andrés, Markus P. Fülscher, Björn O. Roos, and Manuela
Merchán.
Theoretical study of the electronic spectrum of imidazole.
J. Phys. Chem., 100:6484–6491, 1996.
- 261
-
Luis Serrano-Andrés.
Estudio teórico del espectro electrónico de sistemas
orgánicos.
PhD thesis, Universitat de València, 1994.
- 262
-
Luis Serrano-Andrés, Manuela Merchán, Markus Fülscher, and
Björn O. Roos.
A theoretical study of the electronic spectrum of thiophene.
Chem. Phys. Letters, 211:125–134, 1993.
- 263
-
Karl Kaufmann, Werner Baumeister, and Martin Jungen.
Universal Gaussian basis sets for an optimum representation of
Rydberg and continuum wavefunctions.
J. Phys. B: At. Mol. Opt. Phys., 22:2223–2240, 1989.
- 264
-
M. P. Fülscher and B. O. Roos.
The excited states of pyrazine: A basis set study.
Theor. Chim. Acta, 87:403–413, 1994.
- 265
-
Kerstin Andersson.
Multiconfigurational perturbation theory.
PhD thesis, Lunds Universitet, 1992.
- 266
-
Markus P. Fülscher, Luis Serrano-Andrés, and Björn O. Roos.
A theoretical study of the electronic spectra of adenine and guanine.
J. Am. Chem. Soc., 119:6168–6176, 1997.
- 267
-
Luis Serrano-Andrés and Markus P. Fülscher.
Theoretical study of the electronic spectroscopy of peptides. 1.
The peptidic bond: Primary, secondary, and tertiary amides.
J. Am. Chem. Soc., 118:12190–12199, 1996.
- 268
-
Manuela Merchán, Enrique Ortí, and Björn O. Roos.
Theoretical determination of the electronic spectrum of free base
porphin.
Chem. Phys. Letters, 226:27–37, 1994.
- 269
-
Luis Serrano-Andrés and Björn O. Roos.
A theoretical study of the indigoid dyes and their chromophore.
Chem. Eur. J., 3:717–725, 1997.
- 270
-
K. Pierloot, E. Van Praet, L. G. Vanquickenborne, and B. O. Roos.
Systematic ab initio study of the ligand field spectra of
hexacyanometalate complexess.
J. Phys. Chem., 97:12220–12228, 1993.
- 271
-
Kristine Pierloot, Jan O. A. De Kerpel, Ulf Ryde, and Björn O. Roos.
Theoretical study of the electronic spectrum of plastocyanin.
J. Am. Chem. Soc., 119:218–226, 1997.
- 272
-
Kristine Pierloot, Eftimios Tsokos, and Björn O. Roos.
3p–3d intershell correlation effects in transition metal ions.
Chem. Phys. Letters, 214:583–590, 1993.
- 273
-
Manuela Merchán and Remedios González-Luque.
Ab initio study on the low-lying excited states of retinal.
J. Chem. Phys., 106:1112–1122, 1997.
- 274
-
Luis Serrano-Andrés, Manuela Merchán, Björn O. Roos, and Roland
Lindh.
Theoretical study of the internal charge transfer in
aminobenzonitriles.
J. Am. Chem. Soc., 117:3189–3204, 1995.
- 275
-
Manuela Merchán, Rosendo Pou-Amérigo, and Björn O. Roos.
A theoretical study of the dissociation energy of 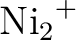 . A
case of broken symmetry. . A
case of broken symmetry.
Chem. Phys. Letters, 252:405–414, 1996.
- 276
-
M. P. Fülscher, S. Matzinger, and T. Bally.
Excited states in polyene radical cations. An ab initio theoretical
study.
Chem. Phys. Letters, 236:167–176, 1995.
- 277
-
Mercedes Rubio, Manuela Merchán, Enrique Ortí, and Björn O. Roos.
A theoretical study of the electronic spectra of the biphenyl cation
and anion.
J. Phys. Chem., 99:14980, 1995.
- 278
-
Vincenzo Barone and Maurizio Cossi.
Quantum calculation of molecular energies and energy gradients in
solution by a conductor solvent model.
J. Phys. Chem. A, 102:1995–2001, 1998.
- 279
-
Maurizio Cossi, Nadia Rega, Giovanni Scalmani, and Vincenzo Barone.
Polarizable dielectric model of solvation with inclusion of charge
penetration effects.
J. Chem. Phys., 114:5691–5701, 2001.
- 280
-
Gunnar Karlström.
New approach to the modeling of dielectric media effects in ab initio
quantum chemical calculations.
J. Phys. Chem., 92:1315–1318, 1988.
- 281
-
Luis Serrano-Andrés, Markus P. Fülscher, and Gunnar Karlström.
Solvent effects on electronic spectra studied by multiconfigurational
perturbation theory.
Int. J. Quantum Chem., 65:167–181, 1997.
- 282
-
Jacopo Tomasi and Maurizio Persico.
Molecular interactions in solution: An overview of methods based on
continuous distributions of the solvent.
Chem. Rev., 94:2027–2094, 1994.
- 283
-
Maurizio Cossi and Vincenzo Barone.
Solvent effect on vertical electronic transitions by the polarizable
continuum model.
J. Chem. Phys., 112:2427–2435, 2000.
- 284
-
Anders Bernhardsson, Roland Lindh, Gunnar Karlström, and Björn O. Roos.
Direct self-consistent reaction field with Pauli repulsion:
Solvation effects on methylene peroxide.
Chem. Phys. Letters, 251:141–149, 1996.
- 285
-
W. F. Forbes and R. Shilton.
Electronic spectra and molecular dimensions. III. Steric effects
in methyl-substituted 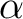 , ,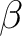 -unsaturated aldehydes. -unsaturated aldehydes.
J. Am. Chem. Soc., 81:786–790, 1959.
- 286
-
Marvin Douglas and Norman M. Kroll.
Quantum electrodynamical corrections to the fine structure of helium.
Ann. Phys., 82:89–155, 1974.
- 287
-
Bernd A. Hess.
Relativistic electronic-structure calculations employing a
two-component no-pair formalism with external-field projection operators.
Phys. Rev. A, 33:3742–3748, 1986.
- 288
-
Per-Åke Malmqvist, Björn O. Roos, and Bernd Schimmelpfennig.
The restricted active space (RAS) state interaction approach with
spin–orbit coupling.
Chem. Phys. Letters, 357:230–240, 2002.
- 289
-
Bernd A. Heß, Christel M. Marian, Ulf Wahlgren, and Odd Gropen.
A mean-field spin–orbit method applicable to correlated
wavefunctions.
Chem. Phys. Letters, 251:365–371, 1996.
- 290
-
B. Schimmelpfennig.
AMFI, an atomic mean-field spin–orbit integral program.
Computer code, 1996.
University of Stockholm.
- 291
-
Björn O. Roos and Per-Åke Malmqvist.
On the effects of spin–orbit coupling on molecular properties:
Dipole moment and polarizability of 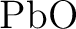 and spectroscopic constants
for the ground and excited states. and spectroscopic constants
for the ground and excited states.
Adv. Quantum Chem., 47:37–49, 2004.
- 292
-
Ulf Wahlgren.
The effective core potential method.
In Björn O. Roos, editor, Lecture Notes in Quantum
Chemistry. European Summer School in Quantum Chemistry, volume 58 of Lecture Notes in Chemistry, pages 413–421. Springer-Verlag, Berlin,
Germany, 1992.
- 293
-
Luis Seijo and Zoila Barandiarán.
The Ab Initio model potential method: A common strategy for
effective core potential and embedded cluster calculations.
In Jerzy Leszczynski, editor, Computational Chemistry: Reviews
of Current Trends, volume 4, pages 55–152. World Scientific, Singapore,
1999.
- 294
-
A. D. Buckingham.
Permanent and induced molecular moments and long-range intermolecular
forces.
Adv. Chem. Phys., 12:107–142, 1967.
- 295
-
Guido Raos, Joseph Gerratt, David L. Cooper, and Mario Raimondi.
Spin correlation in -electron systems from spin-coupled
wavefunctions. I. Theory and first applications.
Chem. Phys., 186:233–250, 1994.
- 296
-
Guido Raos, Joseph Gerratt, David L. Cooper, and Mario Raimondi.
Spin correlation in -electron systems from spin-coupled
wavefunctions. II. Further applications.
Chem. Phys., 186:251–273, 1994.
- 297
-
R. Fletcher.
A new approach to variable metric algorithms.
Comput. J., 13:317–322, 1970.
- 298
-
Gunnar Karlström, Roland Lindh, Per-Åke Malmqvist, Björn O. Roos,
Ulf Ryde, Valera Veryazov, Per-Olof Widmark, Maurizio Cossi, Bernd
Schimmelpfennig, Pavel Neogrády, and Luis Seijo.
MOLCAS: a program package for computational chemistry.
Comput. Mater. Sci., 28:222–239, 2003.
- 299
-
G. A. Gallup and J. M. Norbeck.
Population analyses of valence-bond wavefunctions and 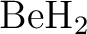 . .
Chem. Phys. Letters, 21:495–500, 1973.
- 300
-
Thomas A. Halgren and William N. Lipscomb.
The synchronous-transit method for determining reaction pathways and
locating molecular transition states.
Chem. Phys. Letters, 50:225–232, 1977.
- 301
-
Ola Engkvist, Per-Olof Åstrand, and Gunnar Karlström.
Accurate intermolecular potentials obtained from molecular wave
functions: Bridging the gap between quantum chemistry and molecular
simulations.
Chem. Rev., 100:4087–4108, 2000.
- 302
-
Isaiah Shavitt.
Matrix element evaluation in the unitary group approach to the
electron correlation problem.
Int. J. Quantum Chem., 14-S12:5–32, 1978.
- 303
-
David L. Cooper, Robert Ponec, Thorstein Thorsteinsson, and Guido Raos.
Pair populations and effective valencies from ab initio SCF and
spin-coupled wave functions.
Int. J. Quantum Chem., 57:501–518, 1996.
- 304
-
Thorstein Thorsteinsson and David L. Cooper.
Modern valence bond descriptions of molecular excited states: An
application of CASVB.
Int. J. Quantum Chem., 70:637–650, 1998.
- 305
-
Ulf Ryde and Mats H. M. Olsson.
Structure, strain, and reorganization energy of blue copper models in
the protein.
Int. J. Quantum Chem., 81:335–347, 2001.
- 306
-
Valera Veryazov, Per-Olof Widmark, Luis Serrano-Andrés, Roland Lindh, and
Björn O. Roos.
MOLCAS as a development platform for quantum chemistry software.
Int. J. Quantum Chem., 100:626–635, 2004.
- 307
-
Ulf Ryde, Mats H. M. Olsson, Björn O. Roos, Jan O. A. De Kerpel, and
Kristine Pierloot.
On the role of strain in blue copper proteins.
J. Biol. Inorg. Chem., 5:565–574, 2000.
- 308
-
Per E. M. Siegbahn, Jan Almlöf, J. Heiberg, and Björn O. Roos.
The complete active space SCF (CASSCF) method in a
Newton–Raphson formulation with application to the HNO molecule.
J. Chem. Phys., 74:2384–2396, 1981.
- 309
-
C. G. Broyden.
The convergence of a class of double-rank minimization algorithms 2.
The new algorithm.
J. Inst. Math. Appl., 6:222–231, 1970.
- 310
-
Ulf Ryde, Mats H. M. Olsson, Kristine Pierloot, and Björn O. Roos.
The cupric geometry of blue copper proteins is not strained.
J. Mol. Biol., 261:586–596, 1996.
- 311
-
Thorstein Thorsteinsson and David L. Cooper.
Nonorthogonal weights of modern VB wavefunctions. Implementation
and applications within CASVB.
J. Math. Chem., 23:105–106, 1998.
- 312
-
Giovanni Li Manni, Simon D. Smart, and Ali Alavi.
Combining the complete active space self-consistent field method and
the full configuration interaction quantum Monte Carlo within a
super-CI framework, with application to challenging metal-porphyrins.
J. Chem. Theory Comput., 12:1245–1258, 2016.
- 313
-
Giovanni Li Manni and Ali Alavi.
Understanding the mechanism stabilizing intermediate spin states in
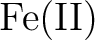 -porphyrin. -porphyrin.
J. Phys. Chem. A, 122(22):4935–4947, 2018.
- 314
-
Giovanni Li Manni, Daniel Kats, David P. Tew, and Ali Alavi.
Role of valence and semicore electron correlation on spin gaps in
-porphyrins.
J. Chem. Theory Comput., 15:1492–1497, 2019.
- 315
-
George H. Booth, Alex J. W. Thom, and Ali Alavi.
Fermion Monte Carlo without fixed nodes: A game of life,
death and annihilation in Slater determinant space.
J. Chem. Phys., 131:054106, 2009.
- 316
-
Catherine Overy, George H. Booth, N. S. Blunt, James J. Shepherd, Deidre
Cleland, and Ali Alavi.
Unbiased reduced density matrices and electronic properties from full
configuration interaction quantum Monte Carlo.
J. Chem. Phys., 141:244117, 2014.
- 317
-
Donald Goldfarb.
A family of variable-metric methods derived by variational means.
Math. Comput., 24:23–26, 1970.
- 318
-
D. F. Shanno.
Conditioning of quasi-Newton methods for function minimization.
Math. Comput., 24:647–656, 1970.
- 319
-
M. J. D. Powell.
Recent advances in unconstrained optimization.
Math. Program., 1:26–57, 1971.
- 320
-
Ove Christiansen, Jürgen Gauss, and Bernd Schimmelpfennig.
Spin–orbit coupling constants from coupled-cluster response theory.
Phys. Chem. Chem. Phys., 2:965–971, 2000.
- 321
-
B. H. Chirgwin and C. A. Coulson.
The electronic structure of conjugated systems. VI.
Proc. Roy. Soc. Lond. A, 201:196–209, 1950.
- 322
-
J. E. Dennis, Jr. and R. B. Schnabel.
Least change secant updates for quasi-Newton methods.
SIAM Rev., 21:443–459, 1979.
- 323
-
Thorstein Thorsteinsson, David L. Cooper, Joseph Gerratt, and Mario Raimondi.
Symmetry adaptation and the utilization of point group symmetry in
valence bond calculations, including CASVB.
Theor. Chim. Acta, 95:131–150, 1997.
- 324
-
Martin Schütz and Roland Lindh.
An integral direct, distributed-data, parallel MP2 algorithm.
Theor. Chim. Acta, 95:13–34, 1997.
- 325
-
H. Bernhard Schlegel.
Optimization of equilibrium geometries and transition structures.
In K. P. Lawley, editor, Ab Initio Methods in Quantum Chemistry
Part I, volume 67 of Advances in Chemical Physics, pages 249–286.
John Wiley & Sons, Hoboken, NJ, USA, 1987.
- 326
-
R. Fletcher.
Practical Methods of Optimization.
John Wiley & Sons, Chichester, West Sussex, England, 2nd edition,
1987.
- 327
-
Warren J. Hehre.
Practical Strategies for Electronic Structure Calculations.
Wavefunction, Irvine, CA, USA, 1995.
- 328
-
Francesco Aquilante, Jochen Autschbach, Rebecca K. Carlson, Liviu F. Chibotaru,
Mickaël G. Delcey, Luca De Vico, Ignacio Fdez. Galván, Nicolas
Ferré, Luis Manuel Frutos, Laura Gagliardi, Marco Garavelli, Angelo
Giussani, Chad E. Hoyer, Giovanni Li Manni, Hans Lischka, Dongxia Ma,
Per Åke Malmqvist, Thomas Müller, Artur Nenov, Massimo Olivucci,
Thomas Bondo Pedersen, Daoling Peng, Felix Plasser, Ben Pritchard, Markus
Reiher, Ivan Rivalta, Igor Schapiro, Javier Segarra-Martí, Michael
Stenrup, Donald G. Truhlar, Liviu Ungur, Alessio Valentini, Steven
Vancoillie, Valera Veryazov, Victor P. Vysotskiy, Oliver Weingart, Felipe
Zapata, and Roland Lindh.
Molcas 8: New capabilities for multiconfigurational quantum
chemical calculations across the periodic table.
J. Comput. Chem., 37(5):506–541, 2016.
- 329
-
Alessio Valentini, Daniel Rivero, Felipe Zapata, Cristina García-Iriepa,
Marco Marazzi, Raúl Palmeiro, Ignacio Fdez. Galván, Diego Sampedro,
Massimo Olivucci, and Luis Manuel Frutos.
Optomechanical control of quantum yield in Trans–Cis
ultrafast photoisomerization of a retinal chromophore model.
Angew. Chem. Int. Ed., 56(14):3842–3846, 2017.
- 330
-
Patrick Kimber and Felix Plasser.
Toward an understanding of electronic excitation energies beyond the
molecular orbital picture, 2020.
- 331
-
S. Knecht, S. Keller, J. Autschbach, and M. Reiher.
A nonorthogonal state-interaction approach for matrix product state
wave functions.
J. Chem. Theory Comput., 12:5881–5894, 2016.
- 332
-
S. Keller, M. Dolfi, M. Troyer, and M. Reiher.
An efficient matrix product operator representation of the
quantum-chemical Hamiltonian.
J. Chem. Phys., 143:244118, 2015.
- 333
-
S. Keller and M. Reiher.
Spin-adapted matrix product states and operators.
J. Chem. Phys., 144:134101, 2016.
- 334
-
S. Knecht, E. D. Hedegård, S. Keller, A. Kovyrshin, Y. Ma, A. Muolo, C. J.
Stein, and M. Reiher.
New approaches for ab initio calculations of molecules with strong
electron correlation.
Chimia, 70:244–251, 2016.
- 335
-
A. A. Granovsky.
Extended multi-configuration quasi-degenerate perturbation theory:
The new approach to multi-state multi-reference perturbation theory.
J. Chem. Phys., 134:214113, 2011.
- 336
-
T. Shiozaki, W. Győrffy, P. Celani, and H.-J. Werner.
Communication: Extended multi-state complete active space
second-order perturbation theory: Energy and nuclear gradients.
J. Chem. Phys., 135:081106, 2011.
- 337
-
Per Åke Malmqvist and Valera Veryazov.
The binatural orbitals of electronic transitions.
Mol. Phys., 110(19-20):2455–2464, 2012.
- 338
-
Attila Szabo and Neil S. Ostlund.
Modern Quantum Chemistry.
McGraw-Hill, New York, NY, USA, 1989.
- 339
-
S. Battaglia and R. Lindh.
Extended dynamically weighted CASPT2: The best of two worlds.
J. Chem. Theory Comput., 16:1555–1567, 2020.
- 340
-
Gerardo Raggi, Ignacio Fdez. Galván, Christian L. Ritterhoff, Morgane
Vacher, and Roland Lindh.
Restricted-variance molecular geometry optimization based on
gradient-enhanced Kriging.
J. Chem. Theory Comput., 16:3989–4001, 2020.
Next: List of Figures
Up: manual
Previous: 10.9 Core and Embedding Potentials
|
|
|







