
























MOLCAS manual: Next: 6. Program Based Tutorials Up: 5. Problem Based Tutorials Previous: 5.2 Optimizing geometries
5.3 Computing excited statesThe calculation of electronic excited states is typically a multiconfigurational problem, and therefore it should preferably be treated with multiconfigurational methods such as CASSCF and CASPT2. We can start this section by computing the low-lying electronic states of the acrolein molecule at the CASSCF level and using a minimal basis set. The standard file with cartesian coordinates is:
We shall carry out State-Averaged (SA) CASSCF calculations, in which one single set of molecular orbitals is used to compute all the states of a given spatial and spin symmetry. The obtained density matrix is the average for all states included, although each state will have its own set of optimized CI coefficients. Different weights can be considered for each of the states, but this should not be used except in very special cases by experts. It is better to let the CASPT2 method to handle that. The use of a SA-CASSCF procedure has an great advantage. For example, all states in a SA-CASSCF calculation are orthogonal to each other, which is not necessarily true for state specific calculations. Here, we shall include five states of singlet character the calculation. As no symmetry is invoked all the states belong by default to the first symmetry, including the ground state.
We have used as active all the
In GRID_IT input we have included all orbitals. It is, however, possible and in general recommended to restrict the calculation to certain sets of orbitals. How to do this is described in the input manual for GRID_IT. Simple math operations can be performed with grids of the same size, for example, LUSCUS can be used to display the difference between two densities. CASSCF wave functions are typically good enough, but this is not the case for electronic energies, and the dynamic correlation effects have to be included, in particular here with the CASPT2 method. The proper input is prepared, again including SEWARD and RASSCF (unnecessary if they were computed previously), adding a CASPT2 input with the keyword MultiState set to 5 1 2 3 4 5. The CASPT2 will perform four consecutive single-state (SS) CASPT2 calculations using the SA-CASSCF roots computed by the RASSCF module. At the end, a multi-state CASPT2 calculation will be added in which the five SS-CASPT2 roots will be allowed to interact. The final MS-CASPT2 solutions, unlike the previous SS-CASPT2 states, will be orthogonal. The FROZen keyword is put here as a reminder. By default the program leaves the core orbitals frozen.
Apart from energies and state properties it is quite often necessary to compute
state interaction properties such as transition dipole moments, Einstein coefficients,
and many other. This can be achieved with the RASSI module, a powerful
program which can be used for many purposes
(see section
Oscillator strengths for the computed transitions and Einstein coefficients are
compiled at the end of the RASSI output file. To obtain these values,
however, energy differences have been used which are obtained from the previous
CASSCF calculation. Those energies are not accurate because they do not include
dynamic correlation energy and it is better to substitute them by properly
computed values, such those at the CASPT2 level. This is achieved with the
keyword Ejob.
More information is available
in section Now a more complex case. We want to compute vertical singlet-triplet gaps from the singlet ground state of acrolein to different, up to five, triplet excited states. Also, interaction properties are requested. Considering that the spin multiplicity differs from the ground to the excited states, the spin Hamiltonian has to be added to our calculations and the RASSI program takes charge of that. It is required first, to add in the SEWARD input the keyword AMFI, which introduces the proper integrals required, and to the RASSI input the keyword SpinOrbit. Additionally, as we want to perform the calculation sequentially and RASSI will read from two different wave function calculations, we need to perform specific links to save the information. The link to the first CASPT2 calculation will saved in file $Project.JobMix.S the data from the CASPT2 result of the ground state, while the second link before the second CASPT2 run will do the same for the triplet states. Later, we link these files as JOB001 and JOB002 to become input files for RASSI. In the RASSI input NrofJobIph will be set to two, meaning two JobIph or JobMix files, the first containing one root (the ground state) and the second five roots (the triplet states). Finally, we have added EJob, which will read the CASPT2 (or MS-CASPT2) energies from the JobMix files to be incorporated to the RASSI results. The magnitude of properties computed with spin-orbit coupling (SOC) depends strongly on the energy gap, and this has to be computed at the highest possible level, such as CASPT2.
As here with keyword AMFI,
when using command Coord to build a SEWARD input
and we want to introduce other keywords, it is enough if we place them
after the line corresponding to Coord.
Observe that the nature of the triplet states obtained is in sequence one
In many cases working with symmetry will help us to perform calculations in quantum chemistry. As it is a more complex and delicate problem we direct the reader to the examples section in this manual. However, we include here two inputs that can help the beginners. They are based on trans-1,3-butadiene, a molecule with a C2h ground state. If we run the next input, the SEWARD and SCF outputs will help us to understand how orbitals are classified by symmetry, whereas reading the RASSCF output the structure of the active space and states will be clarified.
Using the next input will give information about states of a different symmetry. Just run it as a simple exercise.
Structure optimizations can be also performed at the CASSCF, RASSCF or CASPT2 levels. Here we shall optimize the second singlet state in the first (here the only) symmetry for acrolein at the SA-CASSCF level. It is strongly recommended to use the State-Average option and avoid single state CASSCF calculations for excited states. Those states are non-orthogonal with the ground state and are typically heavily contaminated. The usual set of input commands will be prepared, with few changes. In the RASSCF input two states will be simultaneously computed with equal weight (CiRoot 2 2 1), but, in order to get accurate gradients for a specific root (not an averaged one), we have to add Rlxroot and set it to two, which is, among the computed roots, that we want to optimize. The proper density matrix will be stored. The MCLR program optimizes, using a perturbative approach, the orbitals for the specific root (instead of using averaged orbitals), but the program is called automatically and no input is needed.
In case of performing a CASPT2 optimization for an excited state, still the SA-CASSCF approach can be used to generate the reference wave function, but keyword Rlxroot and the use of the MCLR program are not necessary, because CASPT2 takes care of selecting the proper root (the last one). A very useful tool recently included in MOLCAS is the possibility to compute minimum energy paths (MEP), representing steepest descendant minimum energy reaction paths which are built through a series of geometry optimizations, each requiring the minimization of the potential energy on a hyperspherical cross section of the PES centered on a given reference geometry and characterized by a predefined radius. One usually starts the calculation from a high energy reference geometry, which may correspond to the Franck-Condon (FC) structure on an excited-state PES or to a transition structure (TS). Once the first lower energy optimized structure is converged, this is taken as the new hypersphere center, and the procedure is iterated until the bottom of the energy surface is reached. Notice that in the TS case a pair of steepest descent paths, connecting the TS to the reactant and product structures (following the forward and reverse orientation of the direction defined by the transition vector) provides the minimum energy path (MEP) for the reaction. As mass-weighted coordinates are used by default, the MEP coordinate corresponds to the so-called Intrinsic Reaction Coordinates (IRC). We shall compute here the MEP from the FC structure of acrolein along the PES of the second root in energy at the CASSCF level. It is important to remember that the CASSCF order may not be accurate and the states may reverse orders at higher levels such as CASPT2.
As observed, to prepare the input for the MEP is simple, just add the keyword MEP-search and specify a step size with MEPStep, and the remaining structure equals that of a geometry optimization. The calculations are time consuming, because each point of the MEP (four plus the initial one obtained here) is computed through a specific optimization. A file named $Project.mep.molden (read by MOLDEN ) will be generated in $WorkDir containing only those points belonging to the MEP. We shall now show how to perform geometry optimizations under nongeometrical restrictions, in particular, how to compute hypersurface crossings, which are key structures in the photophysics of molecules. We shall get those points as minimum energy crossing points in which the energy of the highest of the two states considered is minimized under the restriction that the energy difference with the lowest state should equal certain value (typically zero). Such point can be named a minimum energy crossing point (MECP). If a further restriction is imposed, like the distance to a specific geometry, and several MECP as computed at varying distances, it is possible to obtain a crossing seam of points where the energy between the two states is degenerated. Those degeneracy points are funnels with the highest probability for the energy to hop between the surfaces in internal conversion or intersystem crossing photophysical processes. There are different possibilities. A crossing between states of the same spin multiplicity and spatial symmetry is named a conical intersection. Elements like the nonadiabatic coupling terms are required to obtain them strictly, and they are not computed presently by MOLCAS. If the crossing occurs between states of the same spin multiplicity and different spatial symmetry or between states of different spin multiplicity, the crossing is an hyperplane and its only requirement is the energetic degeneracy and the proper energy minimization. Here we include an example with the crossing between the lowest singlet (ground) and triplet states of acrolein. Notice that two different states are computed, first by using RASSCF to get the wave function and then ALASKA to get the gradients of the energy. Nothing new on that, just the information needed in any geometry optimizations. The GATEWAY input requires to add as constraint an energy difference between both states equal to zero. A specific instruction is required after calculating the first state. We have to copy the communication file RUNFILE (at that point contains the information about the first state) to RUNFILE2 to provide later SLAPAF with proper information about both states:
Solvent effects can be also applied to excited states, but first the reaction field in the ground (initial) state has to be computed. This is because solvation in electronic excited states is a non equilibrium situation in with the electronic polarization effects (fast part of the reaction field) have to treated apart (they supposedly change during the excitation process) from the orientational (slow part) effects. The slow fraction of the reaction field is maintained from the initial state and therefore a previous calculation is required. From the practical point of view the input is simple as illustrated in the next example. First, the proper reaction-field input is included in SEWARD, then a RASSCF and CASPT2 run of the ground state, with keyword RFPErt in CASPT2, and after that another SA-CASSCF calculation of five roots to get the wave function of the excited states. Keyword NONEequilibrium tells the program to extract the slow part of the reaction field from the previous calculation of the ground state (specifically from the JOBOLD file, which may be stored for other calculations) while the fast part is freshly computed. Also, as it is a SA-CASSCF calculation (if not, this is not required) keyword RFRoot is introduced to specify for which of the computed roots the reaction field is generated. We have selected here the fifth root because it has a very large dipole moment, which is also very different from the ground state dipole moment. If you compare the excitation energy obtained for the isolated and the solvated system, a the large red shift is obtained in the later.
A number of simple examples as how to proceed with the most frequent quantum chemical problems computed with MOLCAS have been given above. Certainly there are many more possibilities in MOLCAS 8.1 such as calculation of 3D band systems in solids at a semiempirical level, obtaining valence-bond structures, the use of QM/MM methods in combination with a external MM code, the introduction of external homogeneous or non homogeneous perturbations, generation of atomic basis sets, application of different localization schemes, analysis of first order polarizabilities, calculation of vibrational intensities, analysis, generation, and fitting of potentials, computation of vibro-rotational spectra for diatomic molecules, introduction of relativistic effects, etc. All those aspects are explained in the manual and are much more specific. Next section 6 details the basic structure of the inputs, program by program, while easy examples can also be found. Later, another chapter includes a number of extremely detailed examples with more elaborated quantum chemical examples, in which also scientific comments are included. Examples include calculations on high symmetry molecules, geometry optimizations and Hessians, computing reaction paths, high quality wave functions, excited states, solvent models, and computation of relativistic effects.
Next: 6. Program Based Tutorials Up: 5. Problem Based Tutorials Previous: 5.2 Optimizing geometries |
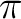 and
and 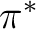 orbitals, two bonding and
two antibonding
orbitals, two bonding and
two antibonding 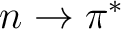 states, and two
states, and two 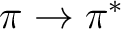 states.
It is convenient to add the GRID_IT input in order to be able to use
the LUSCUS interface for the analysis of the orbitals and the occupations
in the different electronic states. Such an analysis should always be made in
order to understand the nature of the different excited states.
In order to get a more detailed analysis of the nature of the obtained states it is
also possible to obtain in a graphical way the charge density differences between
to states, typically the difference between the ground and an excited state. The
following example creates five different density files:
states.
It is convenient to add the GRID_IT input in order to be able to use
the LUSCUS interface for the analysis of the orbitals and the occupations
in the different electronic states. Such an analysis should always be made in
order to understand the nature of the different excited states.
In order to get a more detailed analysis of the nature of the obtained states it is
also possible to obtain in a graphical way the charge density differences between
to states, typically the difference between the ground and an excited state. The
following example creates five different density files:
![[*]](crossref.png)
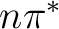 , two
, two 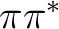 , and two
, and two 
