MOLCAS manual:
Next:
6.2 Environment and EMIL Commands
Up:
6. Program Based Tutorials
Previous:
6. Program Based Tutorials
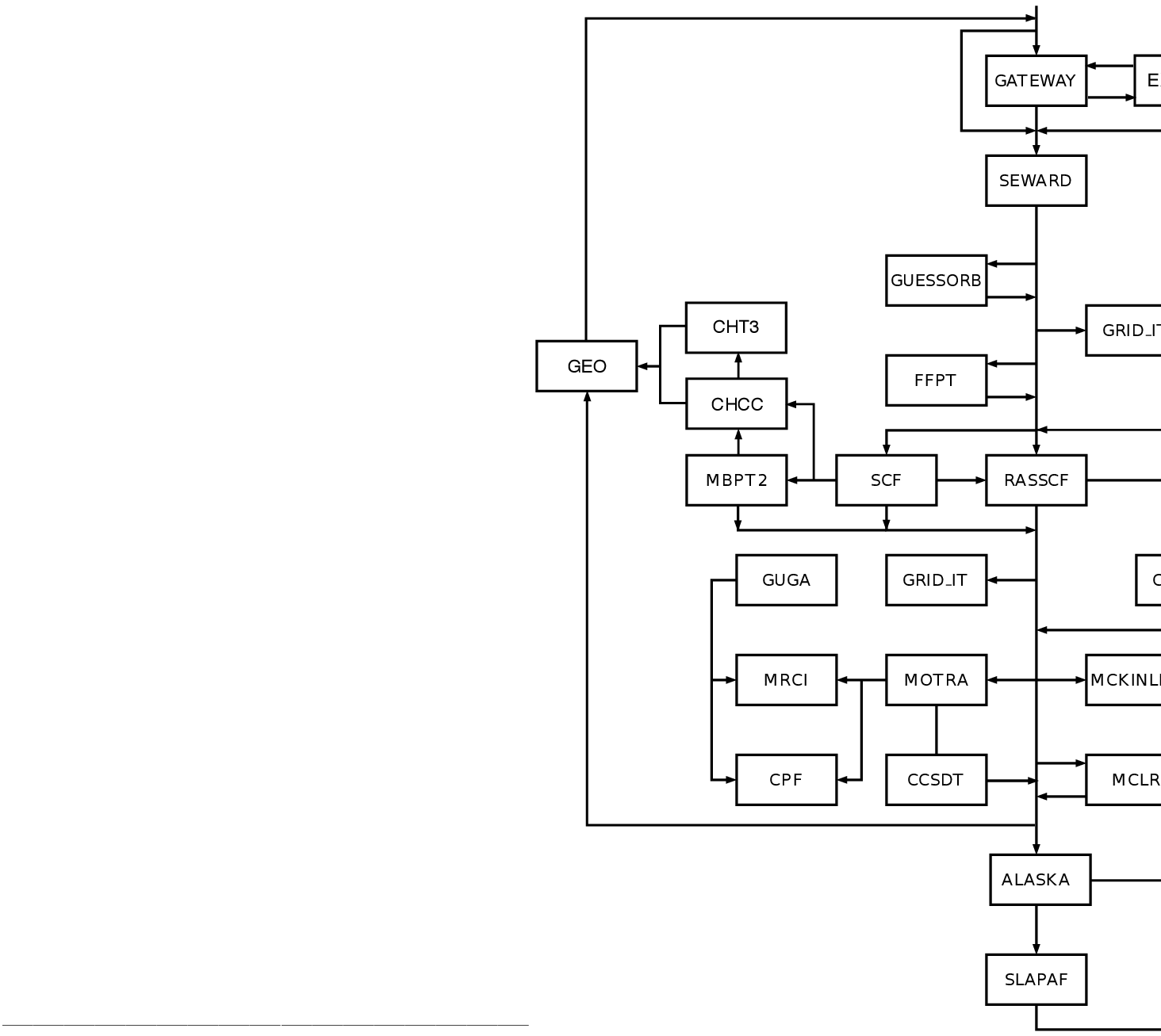
6.1 8.1 Flowchart
Figure:
Flowchart for Module Dependencies in MOLCAS
Next:
6.2 Environment and EMIL Commands
Up:
6. Program Based Tutorials
Previous:
6. Program Based Tutorials