MOLCAS manual:

Next: 6.28 Some practical HINTS
Up: 6. Program Based Tutorials
Previous: 6.26 Most frequent error messages
6.27 Tools for selection of the active space
Selecting an active space is sometimes easy. For a small molecule,
an active space for the ground and the lowest valence excited states
is usually the valence orbitals, i.e. orbitals composed of atomic
orbitals belonging to the usual 'valence shells' (there are
some exceptions to this rule). Problems arise for medium or large
molecules, for higher excited states, and for molecules including
transition, lanthanide or actinide elements. A good wish list
of orbitals will give a CASSCF/CASPT2 calculation that demand
unrealistically large computer resources and time.
Compromises must be made. Any smaller selection of active orbitals
can in general affect your results, and the selection should be
based on the specific calculations: see 6.27 for
advise.
The following three tools may be help in the process:
- localisation
- is a program that can take a (subrange of) orbitals
from an orbital file, and produce a new orbital file where these orbitals
have been transformed to become localized, while spanning the same space
as the original ones.
- expbas
- can take an orbital file using a smaller basis set, and
'expand' it into a new orbital file using a larger basis.
- LUSCUS
- (is of course also described elsewhere) is the orbital viewer.
It is of course best to have a good perception of the electronic structure
of the molecule, including all states of interest for the calculation.
If it is a larger system, where lots of ligands can be assumed not to
partake in non-dynamic correlation, it is a good idea to run some simple
exploratory calculations with a much smaller model system.
Check the literature for calculations on similar systems or model systems.
First of all, you need to know how many orbitals (in each symmetry) that
should be active. Their precise identity is also good to know, in order
to have a good set of starting orbitals, but we come to that later.
Necessary active orbitals are: Any shells that may be open in any of the
states or structures studied. Breaking a bond generally produces a
correlated bond orbital and a correlating antibonding orbital, that must
both be active (Since it is the number of orbitals we are dealing
with as yet, you may as well think of the two radical orbitals that are
produced by completely breaking the bond).
You probably want to include one orbital for each aromatic carbon.
Valuable correlated active orbitals are: Oxygen lone pair, CC
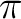 bonds. Valuable correlating active orbitals are: the
antibonding bonds. Valuable correlating active orbitals are: the
antibonding 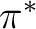 CC orbitals, and one additional set of
correlating d orbitals for most transition elements (sometimes
called the 'double d-shell effect'). CC orbitals, and one additional set of
correlating d orbitals for most transition elements (sometimes
called the 'double d-shell effect').
The valuable correlated orbitals can be used as Ras-1 orbitals, and
correlating ones can be used as Ras-3 orbitals, if the active space
becomes too large for a casscf calculation.
Assuming we can decide on the number of active orbitals, the next task
is to prepare starting orbitals that enables CASSCF to converge, by
energy optimization, to the actual starting orbitals for your calculation.
Use a very small basis set to begin with: This will usually be one of the
minimal bases, e.g. ANO-S-MB. This is not just to save time: the small
basis and the large energy spacings make it much easier to get well-defined
correlating orbitals.
Performing the actual casscf (or rasscf) calculation may give you the
active space you want: Viewing the orbitals by LUSCUS may confirm this, but
very often the orbitals are too mixed up (compared to ones mental
picture of what constitutes the best orbitals).
Using localisation program solves this problem. In order to localise
without mixing up orbitals from different subspaces may require to
produce the new orbital file through several runs of the program;
however, for the present perpose, it may be best not to have so
very strict restrictions, for example: Allow mixing among a few
high inactive and the most occupied orbitals; and also among the
weakly occupied and some virtual orbitals.
Running the localisation program, and viewing the localised orbitals,
is a great help since directly in LUSCUS one can redefine orbitals as
being inactive, or ras3 , or whatever, to produce a new orbital file.
The resulting annotated localised orbitals can be used in a new run.
Once a plausible active space has been found, use the expbas tool to
obtain starting orbitals using, e.g. ANO-VDZP basis, or whatever is
to be used in the bulk of the production run.
It is also a good idea to, at this point, 'waste' a few resources on
a single-point calculation for a few more states than you are really
interested in, and maybe look at properties, etc. There may be
experimental spectra to compare with.
And please have a look at the following
section, 'Some practical HINTS' 6.27.
Next: 6.28 Some practical HINTS
Up: 6. Program Based Tutorials
Previous: 6.26 Most frequent error messages
|


























