
























MOLCAS manual: Next: 6.4 SEWARD An Up: 6. Program Based Tutorials Previous: 6.2 Environment and EMIL Commands
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | Generators | MOLCAS | Elements | |||||||||||
| g1 | g2 | g3 | g1 | g2 | g3 | E | g1 | g2 | g1g2 | g3 | g1g3 | g2g3 | g1g2g3 | |
| C1 | E | |||||||||||||
| C2 | C2 | xy | E | C2 | ||||||||||
| Cs | 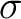 |
x | E | |
||||||||||
| Ci | i | xyz | E | i | ||||||||||
| C2v | C2 | 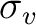 |
xy | y | E | C2 | |
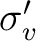 |
||||||
| C2h | C2 | i | xy | xyz | E | C2 | i | 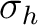 |
||||||
| D2 | C2z | C2y | xy | xz | E | C2z | C2y | C2x | ||||||
| D2h | C2z | C2y | i | xy | xz | xyz | E | C2z | C2y | C2x | i | 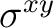 |
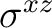 |
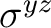 |
To reduce the input, the unity operator E is always assumed. The twofold
rotation about the z-axis, C2(z), and the reflection in the xz-plane,
(xz), are input as XY and Y respectively. The MOLCAS input can be viewed as symmetry operators that operate on the
Cartesian elements specified. For example, the reflection in the
xz-plane is specified by the input keyword Y which is the
Cartesian element operated upon by the reflection.
The input produces the character table in the
GATEWAY section of the output shown in
Figure 6.3. Note that (yz) was produced from
the other two generators. The last column contains the basis functions of
each irreducible symmetry representation. The totally symmetric a1
irreducible representation has the z basis function listed which is unchanged
by any of the symmetry operations.
E C2(z) s(xz) s(yz)
a1 1 1 1 1 z
b1 1 -1 1 -1 x, xz, Ry
a2 1 1 -1 -1 xy, Rz, I
b2 1 -1 -1 1 y, yz, Rx
The geometry of the molecule is defined using the keyword coord. On the next line, the name of the xyz file that defines the geometrical parameters of the molecule (water.xyz) is given.
- The first line of the water.xyz file contains the number of atoms.
- The second line is used to indicate the units: Ångström or atomic units.
The default is to use Ångström.
- Then follows one line for each atom containing the name of each atom and its coordinates.
Basis sets are defined after the keyword BASIs sets. The oxygen and hydrogen basis set chosen, for this example, are the small Atomic Natural Orbitals (ANO) sets. There are three contractions of the basis included in the input, which can be toggled in or excluded with an asterisk, according to the desired calculation: minimal basis, double zeta basis with polarization, or triple zeta basis with polarization.

3 O .000000 .000000 .000000 H 0.700000 .000000 0.700000 H -0.700000 .000000 0.700000
6.3.1 GATEWAY Output
The GATEWAY output contains the symmetry character table, basis set information and input atomic centers. The basis set information lists the exponents and contraction coefficients as well as the type of Gaussian functions (Cartesian, spherical or contaminated) used.
The internuclear distances and valence bond angles (including dihedral angles) are displayed after the basis set information. Inertia and rigid-rotor analysis is also included in the output along with the timing information.
A section of the output that is useful for determining the input to the MOLCAS module SCF is the symmetry adapted basis functions which appears near the end of the GATEWAY portion of the output. This is covered in more detail in the SCF tutorial.
The most important file produced by the GATEWAY module is the RUNFILE which in our case is linked to water.RunFile. This is the general MOLCAS communications file for transferring data between the various MOLCAS program modules. Many of the program modules add data to the RUNFILE which can be used in still other modules. A new RUNFILE is produced every time GATEWAY is run. It should finally be mentioned that for backwards compatibility one can run MOLCAS without invoking GATEWAY. The corresponding input and output will then be handled by the program SEWARD.
6.3.2 Basis Set Superposition Error (BSSE)
GATEWAY can operates with several coordinate files, which is convenient for computing BSSE corrections. BSSE followed by a number marks a XYZ file which should be treated as dummy atoms. The following example demonstrates this feature:
&GATEWAY
coord = ethanol.xyz
coord = water.xyz
bsse = 1
basis = ANO-S-MB
NOMOVE
&SEWARD; &SCF
&GRID_IT
NAME = water
***************
&GATEWAY
coord = ethanol.xyz
coord = water.xyz
bsse = 2
basis = ANO-S-MB
NOMOVE
&SEWARD; &SCF
&GRID_IT
NAME = ethanol
**************
&GATEWAY
coord = ethanol.xyz
coord = water.xyz
basis = ANO-S-MB
NOMOVE
&SEWARD; &SCF
&GRID_IT
NAME = akvavit
Note, that NOMOVE keyword prevents centering of the molecule, so the computed grids are identical. An alternative way to compute density difference is to modify coordinates, and change an element label to X.
6.3.3 GATEWAY Basic and Most Common Keywords
| Keyword | Meaning |
| Coord | File name or inline number of atoms and XYZ coordinates |
| BASIs Set | Atom_label.Basis_label (for example ANO-L-VTZP) |
| Group | Full (find maximum), NoSym, or symmetry generators |
| SYMMetry | Symmetry generators: X, Y, Z, XY, XZ, YZ, XYZ (in native format) |
| RICD | On-the-fly auxiliary basis sets. |
Next: 6.4 SEWARD An Up: 6. Program Based Tutorials Previous: 6.2 Environment and EMIL Commands

