MOLCAS manual:

Next: IV. User's Guide
Up: 6. Program Based Tutorials
Previous: 6.27 Tools for selection of
Subsections
6.28 Some practical HINTS
This section contains a collection of practical hints
and advices how to
use MOLCAS in solving quantum chemistry problems.
- Try the Cholesky approximation (or RI)!
It saves disk space and possibly time.
- Think about basis set. ANO-like basis sets have many advantages,
but they are "marginal".
- Try to avoid inline basis sets, use the library.
- Remember that the quality of basis set should match quality of
computational method.
- Use ANO-RCC even for atoms in the 2nd row.
- Be extremely careful when computing anions.
Remember that special situations requires special basis sets.
- Use minimal or small basis set for understanding chemical problem.
You always can use expbas later..
- HF orbitals are in many cases good starting orbitals,
but quite often GuessOrb can be used instead.
- Very large basis set together with HF can lead to linear dependences.
- Remember! Hartree-Fock method allows multiple solutions (even for trivial
systems)
- Be reasonable selecting convergence thresholds
- UHF convergence is much poor comparing to RHF.
- Sometime you have to use Scramble keyword to break the symmetry.
- DFT convergence can't be better that HF convergence. Think about starting
orbitals for DFT.
- Remember that DFT is a powerful method but
it is still single configurational method. Don't use it beyond
it's limits.
- Choose your favorite functional, and stay with your decision
- Note that MOLCAS is not the best DFT code available
- MCSCF are multi-solution methods that heavily depend on
the starting orbitals and
level of calculation (roots).
- On convergence ALWAYS (ALWAYS, ALWAYS, etc) check the orbitals
(LUSCUS, molden, CMOcorr, etc). MCSCF methods will lead to different solutions
for active spaces of
different nature. Use your chemical intuition and
let the calculation guide you.
- Analyze carefully the CI coefficients and natural occupation
numbers together with
the orbitals (average orbitals are fine in general for that)
in order to understand the
nature of the states .
- You get average orbitals, and orbitals for individual roots, which
you can visualized by LUSCUS or molden
etc, contain the natural orbitals of the different roots.
- Try increasing the number of SA-CASSCF roots to locate more excited states. They can
be low-lying solutions at the CASPT2 level. In high symmetry cases you may also need
to consider roots that have high energy at the initial steps and can become lower roots in
the converged calculation.
- It is NOT advisable to play games with weights for the different roots. Roots with equal
weights make your calculation more clear and reproducible.
- MOLCAS can handle only D2h subgroups. Molecules with other
symmetry (C3v, D4d, Td, Oh) have a problem.
Especially if you use approximations, like CD.
- Work in a symmetry point group that allows degenerate states to belong to the
same irreducible representation (e.g. C2 for linear molecules). Try C1 too.
- Working in a too high symmetry might prevent you from obtaining less
symmetric lowest-lying localized solutions (e.g. Ni2+)
- Start with clean symmetric orbitals (GUESSORB). Sometime (for example
for a radical), an orbital
for positively charged system can be more symmetric.
- use if needed, CLEANUP and SUPSYM, or for linear molecules: the
LINEAR keyword.
- use it! RASSCF is a simple way to increase an active space.
- Balance RAS1/3 and RAS2 subspaces. Try to change orbitals between
these subspaces.
- Removing RAS2 space completely is not a good idea.
- Note that RAS calculations have a slower convergence, and demand more
resources.
- Increase LEVShift parameter in cases of slow or difficult convergence.
- Sometimes RASSCF is very sensitive when is close to convergence.
Try restarting the calculation from the previous JOBIPH file
- Try to restart from orbitals (or JOBIPH) instead of starting from scratch.
- Always compare calculations with the same active space size (and nature if possible).
- Ask yourself first which is your goal. The selection of the active space depends on that.
- If you made a selection once, try to reuse orbitals! Especially for a set of
calculations with different geometries
- In ground state calculations many orbitals can have an occupation close to 2 and 0 and
might rotate with others in the inactive (secondary) space. It might be wise to skip them
- For low-lying excited states and few roots you might leave inactive quite a number of
orbitals. Check with RASSCF for instance.
- SCF orbital energies sometimes help to choose the orbitals by using the energy order
criterion, but you must learn to see the problems (like lone pair orbitals having too low
energies at the SCF level).
- You typically will need correlating orbitals, that is, if you have a
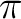 orbital you need a orbital you need a 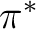 ,
the same for ,
the same for 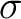 , ,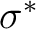 , but not for lone pairs. , but not for lone pairs.
- CASSCF/RASSCF geometry optimizations are the worst case. If you miss orbitals you
might end up in a totally wrong geometry (e.g. breaking a bond usually requires the
bonding and antibonding orbitals in the space).
- Organic (1st row atoms) molecules usually require open shell orbitals,
, , and lone
pairs. If 2nd row atoms are added (S, P, Si, etc) s orbitals enter in action (s bonds are
longer). CH bonds can often be left be inactive.
- Rydberg states require additional diffuse basis sets and specific orbitals in the active
space. Use basis sets centered in the charge center of the positive ion
(consult the manual).
- Transition metal chemistry (1st row) sometimes requires a double d shell description
in the active space.
- Lanthanides have a quite inert 4f shell that must be active together with 5d, 6s (6p).
Actinides 5f, 6d, 7s.
- use expbas! start from minimal basis set, decide the active
space, and expand the basis to "normal". With small basis set you can
clearly identify orbitals.
- use localization! Especially for virtual orbitals.
- expand active space by adding RAS1/3 - give the system a freedom, and see how it
reacts.
- The new IPEA = 0.25 zeroth Hamiltonian is the default.
It particularly improves open shell cases. But there are some cases where
IPEA=0 gives better correlation with experiment.
- Energy differences between different states or situations are only reliable between
calculations with the same active space size and similar reference weights in CASPT2.
- An intruder state (low reference weight in the CASPT2 state) might be informing you
that your active space lacks an important orbital. Check the list of large perturbative
contributions (small denominators combined with large RHS values; check the output)
and also the occupation number of the CASPT2 orbitals.
- For weakly interacting intruder states cases try the IMAGINARY level shift parameter.
Don't use the level shift to reach agreement with experiment!
- For heavy valence-Rydberg mixing cases or for closely degenerated CASPT2 states,
use MS-CASPT2.
- If the MS-CASPT2 description differs a lot from the CASPT2 one,
try to check the
calculation by increasing the active space (introducing angular correlation if possible)
until the result is converged. The "true" solution is typically between both cases
(CASPT2 and MS-CASPT2). If you are suspicious about the MS-CASPT2 result,
better keep the CASPT2 one. It has worked out generally well so far.
- Remember that the program shows first the interaction among the input states and later this description might change.
ALWAYS check the changing order of states.
- For spin-orbit coupling calculations don't forget to include the CASPT2 energies as input
(EJOB or HDIAG keywords) because the results depend on the energy gap. In other cases
having the CASPT2 energies as input will help you to get the right oscillator strength and
Einstein coefficient in the final table.
- If you have degenerate states be sure that the CASPT2 energies are degenerate. If they
are not (which is common) average the energies for the degenerate set (the two
components of E symmetry for example).
- Remember that the spin-orbit coupled results (e.g. TDM) depend on the number of interacting singlet and triplet states included in RASSI.
- Not all methods have analytical derivatives.
- Default thresholds in slapaf are typically too tight. Do not waste computer time!
- Use constrained optimization
- For minima on flat hypersurfaces, such in loosely bound fragments, or in slow convergence
cases you might have to decrease the CUTOFF threshold in ALASKA
- Be careful with the bond angle definition if you are close to a linear bond.
You may have to switch to the LAngle definition
- Don't forget that CASSCF does not include dynamical correlation. In some cases you better
change to DFT or numerical CASPT2 optimizations or, if this is not feasible, may be
preferable to run RASSCF optimizations
- Poor active spaces may lead you to symmetry broken wrong solutions (e.g. a Cs minimum
for water below the true C2v one)
- Poor geometry convergence might be reduced or at least controlled by reducing the initial
trust radius with the MAXSTEP keyword or/and by doing the optimization in Cartesian
coordinates (CARTESIAN)
- In order to obtain localized solutions it might be a good idea to feed the program with a
slightly distorted geometry that helps the method to reach the non symmetric solutions.
Other possibilities are to use an electric field, to add a charge far from the system or use a solvent cavity. In all cases you break symmetry and allow less symmetric situations.
- Linearly interpolated internal coordinates geometries may be a good starting point to locate
a transition state. Use also the useful FindTS command. Sometimes can be wise to compute
a MEP from the TS to prove that it is relevant for the studied reaction path. Try also the new
Saddle approach!
- When locating a CASSCF surface crossing (MECP) ALWAYS compute CASPT2 energies
at that point. The gap between the states can be large at that level. In severe cases you might have to make a scan with CASPT2 to find a better region for the crossing.
- Remember that (so far) MOLCAS does not search for true conical intersections (CIs) but
minimum energy crossing points (MECP) because it lacks NACMEs. Note however that
typically computed minimum energy CIs (MECIs) may not be photochemically relevant
if they are not easily accessible. Barriers have to be computed. Use MEPs!!
- Numerical hessians and optimizations may lead you to bad solutions when different
electronic states are too close. As you move your calculation from the equilibrium geometry
some of the points may belong to other state and corrupt your result. This might be the case
for numerical CASPT2 crossing search. Use then MS-CASPT2 search.
- Remember that SA-RASSCF analytical gradients and SA-CASSCF analytical hessians are
not implemented.
- Be careful with the change of roots and nature along a geometry optimization or MEP.
For example, you start with the state in root 3 (at the CASSCF level) and reach a region
of crossing root 3 and root 2. You may need to change to root 2 for your state.
Not an easy solution (so far).
- Some effects of the solvent are very specific, such as hydrogen bonds, and require to
include explicit solvent molecules. Try adding a first solvent shell (optimized with
molecular mechanics for instance) and then a cavity, for instance with PCM.
- Too small cavity sizes can lead you to unphysical solutions,
even if they seem to match experiment.
- Remember using NonEquilibrium (final state) and RFRoot (SA-CASSCF)
when required
- QM/MM is a much powerful strategy, but it requires experience and knowledge
of the field
Next: IV. User's Guide
Up: 6. Program Based Tutorials
Previous: 6.27 Tools for selection of
|


























