
























MOLCAS manual: Next: 8.32 motra Up: 8. Programs Previous: 8.30 MCPDFT
|
| &MkNemo &End |
| End Of Input |
Example:
&MkNemo &End
.................
End of input
The MkNemo defines transformation as translation,T, or rotation, R, operation in a format:
[ x y z angle]
where the ![$[x\ y\ z]$](img120.png) is a 3D-vector of translation, or the
is a 3D-vector of translation, or the ![$[x,y\ z]$](img121.png) is a 3D-vector of rotation if the angle parameter is presented, and the angle is an optional parameter which is an angle of rotation around this vector in degrees. Generally, translation and rotation operation do not commute, since that the MkNemo first applys transformation from left to right, i.e.: product
is a 3D-vector of rotation if the angle parameter is presented, and the angle is an optional parameter which is an angle of rotation around this vector in degrees. Generally, translation and rotation operation do not commute, since that the MkNemo first applys transformation from left to right, i.e.: product 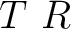 means that the MkNemo will apply first rotation and then translation.
means that the MkNemo will apply first rotation and then translation.
The input of MkNemo module has been split into four groups of keywords:
- Molecules, Clusters, and Displacement,
- GetEenergy,
- Next,
- Test.
| Keyword | Meaning | ||||||||||||
| MOLE, CLUS, and DISP | The keywords must be provided in right order in the input file. And the blocks of keywords, MOLE, CLUS, and DISP, cannot be split between separated MkNemo inputs.
The definition of a Molecule has format:
In the Cluster's block, user defines a cluster in format:
The DISPlacement block contains information about transformations of one of the clusters in the format:
Any atomic coordinates and vectors of transformations must be provided in a.u. units. The coordinates of transformation vector can be separated by space or a comma .Moreover, the Molecule blocks must be provided first, then the Cluster blocks must appear, and finally Displacement block. In a mixed order, the MkNemo will not be able to recognize a label of molecule[cluster] defined below a block which is using it. An execution of MkNemo module within defined Mole, Clus, and Disp blocks in an input will generate a two coordinate files, named MKNEMO.Axyz and MNEMO.Bxyz. Those files contain coordinates of atoms for clusters B and A respectively, and can be used directly in the SEWARD and GATEWAY (see documentation of GATEWAY for COORD keyword). By default, the Seward or Gateway will apply symmetry, so user must be aware that the displacement transformation can break symmetry of the system and the MkNemo does not control it. If you do not want use symmetry see documentation of Seward or Gateway for details.
Example:
| ||||||||||||
| GETE | The GetEnergy block is used to read total energy stored at RUNFILE, and to save it into the MKNEMO.Conf file. The argument of GetEnergy block must be present and it must be a label from the list below. Use
Example:
| ||||||||||||
| NEXT | The Next block is used to save all information about potential curve from previous step into the MKNEMO.Nemo file (the command Next will move data from MKNEMO.Conf file into MKNEMO.Nemo file and will delete MKNEMO.Conf file) and to continue or break an EMIL's loop. This block cannot be used before Mole, Clus, and Disp blocks.
Example:
| ||||||||||||
| TEST | The TEST block CAN BE ONLY USED to save verification data for MOLCAS command verify.
Example:
|
Finally the structure of a standard input file for MkNemo module has the following form:
* Loop over configurations
>>>>>>>>>>>>>>>>>>> Do While <<<<<<<<<<<<<<<<<<<<
&MkNemo&End
* Molecules definitions
Mole : MoleculeName
AtomLabel x y z
......... .. .. ..
AtomLabel x y z
End
....................
Mole : MoleculeName
AtomLabel x y z
......... .. .. ..
AtomLabel x y z
End
*
Clus : ClusterName ClusterTransformation
MoleculeName MoleculeTransformation
............ ......................
MoleculeName
MoleculeName
End
Clus : ClusterName ClusterTransformation
MoleculeName MoleculeTransformation
............
MoleculeName
End
Disp
ClusterName NumberOfSteps [x y z alpha]
ClusterName NumberOfSteps [x y z]
........... ............. .............
ClusterName NumberOfSteps [x y z alpha]
End
End Of Input
*************** SUPER-SYSTEM CALCULATION *********************
* Calculation of integrals
&Seward
coord=$Project.MkNemo.Axyz
coord=$Project.MkNemo.Bxyz
basis=........
................................
* Energy calculation on the first level of the theory
&Scf
...............................
* Save energy
&MkNemo
GetE=S1
* Energy calculation on the second level of the theory
&MBPT2
...............................
* Save energy
&MkNemo
GetE=S2
*************** A-SUBSYSTEM CALCULATION *********************
* Calculation of integrals
&Seward
coord=$Project.MkNemo.Axyz
coord=$Project.MkNemo.Bxyz
* the B-subsytem has charge equal to zero
BSSE=2
basis=........
................................
* Energy calculation on the first level of the theory
&Scf
...............................
* Save energy
&MkNemo&End
GetE=A1
* Energy calculation on the second level of the theory
&MBPT2
...............................
* Save energy
&MkNemo
GetE=A2
*************** B-SUBSYSTEM CALCULATION *********************
* Calculation of integrals
&Seward
coord=$Project.MkNemo.Axyz
coord=$Project.MkNemo.Bxyz
* the A-subsytem has charge equal to zero
BSSE=1
basis=........
................................
* Energy calculation on the first level of the theory
&Scf
...............................
* Save energy
&MkNemo
GetE=B1
* Energy calculation on the second level of the theory
&MBPT2
...............................
* Save energy and take next configuration
&MkNemo
GetE=B2; Next
>>>>>>>>>>>>>>>>>>> EndDo <<<<<<<<<<<<<<<<<<<<
Example:
*
* Loop over all configurations
*
>>>>>>>>>>>>>>>>>>> Do While <<<<<<<<<<<<<<<<<<<<
*
* H2O and H2O clusters
*
&MkNemo&End
* Molecules definitions
Mole : H2O
H 1.43 0.0 1.07
H -1.43 0.0 1.07
O 0.00 0.0 0.00
End
* Clusters definitions
Clus : H2O
H2O : [0.0 1.0 0.0 180.0]
End
Clus : h2o [ 0.0 0.0 2.0]
H2O
End
Disp
h2o : 10 [0.0 0.0, 5.0 ]
h2o : 10 [0.0, 0.0, 20.0 ]
h2o : 18 [0.0 0.0 1.0 180.0]
End
End Of Input
*************** SUPER-SYSTEM CALCULATION *********************
* Calculation of integrals
&Seward
NEMO
Title=Sypersystem
Douglas-Kroll
ANGM= 0.0 0.0 0.0; AMFI
COORD=$Project.MkNemo.Axyz;Coord=$Project.MkNemo.Bxyz
basis=H.ano-rcc...2s1p.,O.ano-rcc.Roos..4s3p2d1f.
* Energy calculation on the first level of the theory
&Scf
Title=Supersystem; Occupied=10; Iterations=30; Disk=1 0
* Save energy
&MkNemo
GetE=S1
* Energy calculation on the second level of the theory
&MBPT2
Title=Sypersystem; Threshold=1.0d-14 1.0d-14 1.0d-14
* Save energy
&MkNemo
GetE=S2
*************** A-SUBSYSTEM CALCULATION *********************
* Calculation of integrals
&Seward
NEMO
Title=A-system
Douglas-Kroll
ANGM= 0.0 0.0 0.0; AMFI
COORD=$Project.MkNemo.Axyz;Coord=$Project.MkNemo.Bxyz
basis=H.ano-rcc...2s1p.,O.ano-rcc.Roos..4s3p2d1f.
BSSE=2
* Energy calculation on the first level of the theory
&Scf
Title=A-subsystem; Occupied=5; Iterations=30; Disk=1 0
* Save energy
&MkNemo
GetE=A1
* Energy calculation on the second level of the theory
&MBPT2
Title=A-subsystem; Threshold=1.0d-14 1.0d-14 1.0d-14
* Save energy
&MkNemo
GetE=A2
*************** B-SUBSYSTEM CALCULATION *********************
* Calculation of integrals
&Seward
NEMO
Title=A-system
Douglas-Kroll
ANGM= 0.0 0.0 0.0; AMFI
COORD=$Project.MkNemo.Axyz;Coord=$Project.MkNemo.Bxyz
basis=H.ano-rcc...2s1p.,O.ano-rcc.Roos..4s3p2d1f.
BSSE=1
* Energy calculation on the first level of the theory
&Scf
Title=B-subsystem; Occupied=5; Iterations=30; Disk=1 0
* Save energy
&MkNemo
GetE=B1
* Energy calculation on the second level of the theory
&MBPT2
Title=B-subsytem; Threshold= 1.0d-14 1.0d-14 1.0d-14
* Save energy and take next configuration
&MkNemo
GetE=B2; Next
>>>>>>>>>>>>>>>>>>> EndDo <<<<<<<<<<<<<<<<<<<<
In this example we calculate potential energy curve for interaction between two water clusters. The A-cluster, H2O, was rotated around Y-axis about 180 degrees. The B-subsystem, h2o,has been translated along Z-axis by 2 a.u.. In the Disp block we have defined 20 translation operations for h2o cluster and 18 rotation operations for H2O cluster. For energy calculations of super-system, A-subsystem, and B-subsystem, at first level of theory we used SCF module, and MBPT2 at second level of theory, respectively. After a calculation of energy we save calculated results using keyword GetE with proper argument in the MKNEMO.Conf file of MkNemo module. Finally, by calling block Next of MkNemo, we save all informations about potential for given configuration and we generate new configuration. This procedure will be repeated for all translations and rotations defined in the Displacement block.
8.31.2.2 Input files
Apart from the standard input unit MkNemo will use the following input files.
| File | Contents | |
| MKNEMO.Input | A MkNemo's input file contains the latest preprocessed input. | |
| MKNEMO.Restart | The MKNEMO.Restart is a restart file, which will be generated by MkNemo at the first run if the file does not exist. Any call of group of command: Mole, Clus, and Disp will be updated and the restart file is saved in user's $CurrDir. If MkNemo calculation crashes, one can fix a reason of crash, copy restart and MKNEMO.Nemo files to $WorkDir, and run the calculation again. The MkNemo will restart calculation from the last point which has been finished successfully. If the MKNEMO.Nemo file will not be copied the MkNemo will generate a new one and will overwrite the file in your $CurrDirr if any exist. Beware of it.
The restart file is formated :
| |
| MKNEMO.Conf | The MKNEMO.Conf is a file which stores block Mole, Clus, and Eneries in similar format like it is define in the input of theMkNemo, but within XML format. The propose of this file is to share definition of molecules, clusters, and energies between different blocks of namelist,
* Configuration definition - contains informations * about configuration <Configuration> * Definition of molecule <Molecule Name=''Name of molecule''> labelOfAtom x y z ........... .. .. .. labelOfAtom x y z </Molecule> .................................... <Molecule Name=''Name of molecule''> labelOfAtom x y z ........... .. .. .. labelOfAtom x y z </Molecule> * Definition of cluster <Cluster Name=''Name of cluster A'' Transformation=''x y z q0 q1 q2 q3''> labelOfMolecule x' y' z' q0' q1' q2' a3' ............... .. .. .. .. .. .. .. labelOfMolecule x' y' z' q0' q1' q2' a3' </Cluster> <Cluster Name=''Name of cluster B'' Transformation=''x y z q0 q1 q2 q3''> labelOfMolecule x' y' z' q0' q1' q2' a3' ............... .. .. .. .. .. .. .. labelOfMolecule x' y' z' q0' q1' q2' a3' </Cluster> * Enerigies definition <Energies> EnegyLabel MethodLabel Energy .......... ........... ...... EnegyLabel MethodLabel Energy </Energies> </Configuration> where the EnergyLabel is one of labels defined in the GetE block, the MethodLabel is a name of method which has been used to calculate energy, and Energy is a vector of eigenvalues. The Next command will save energy information into a MKNEMO.Nemo file, and will clear this file. Hacking hint: If you want to use Rassi then do not use call of command GetE but postprocess output and print eigenvalues to the MKNEMO.Conf file in the right format (use ! in user input to execute shell command for postprocessing of output) |
Files of the SEWARD, SCF, RASSCF, MBPT2, MOTRA, CCSDT, and CASPT2 modules are needed to get total energy on each level of theory for subsystems and super-system.
8.31.2.3 Output files
In addition to the standard output unit MkNemo will generate the following files.
| File | Contents |
| MKNEMO.Axyz,MKNEMO.Bxyz | The MKNEMO.*xyz file is a file of coordinates in format:
NumberOfAtoms AdditionalLine AtomLabel x y z ......... .. .. .. AtomLabel x y z where the NumberOfAtoms is a number of atoms in the file, the AdditionalLine is a line where one can provide unit of coordinate (currently MkNemo supports only a.u.), the AtomLabel is a label of atom, and x, y, z is a vector of coordinates. |
| RUNFILE | A file with informations needed by the block of MOLCAS. |
| MKNEMO.Nemo | On this file MkNemo will store all information about intermolecular potential in the NEMO file format. This format is used by NEMO to fit intermolecular potential to the NEMO model. The format of this file is defined as follows:
<Nemo> * Definition of configuration <Configuration> ............................. </Configuration> ............................. <Configuration> ............................. </Configuration> </Nemo> Where configuration block is defined like in the MKNEMO.Conf.
|
8.31.3 Dependencies
The MkNemo depends on the modules of MOLCAS program, which calculate the total energy of the system.
Next: 8.32 motra Up: 8. Programs Previous: 8.30 MCPDFT

