
























MOLCAS manual: Next: 8.33 mpprop Up: 8. Programs Previous: 8.31 mknemo
|
| File | Contents |
| ONEINT | One-electron integral file generated by SEWARD. |
| ORDINT* | Ordered two-electron integrals generated by SEWARD. |
| INPORB | If MO's are read in formatted form. |
| JOBIPH | If molecular orbitals are read from a RASSCF interface. |
8.32.2.2 Output files
The program MOTRA creates two files: The first carries all basic information and a list of transformed one-electron integrals. The second file includes the transformed two-electron integrals.
The following is a list of output files
| File | Contents |
| TRAONE | Auxiliary data and transformed one-electron integrals. |
| TRAINT* | Transformed two-electron integrals. |
8.32.3 Input
This section describes the input to the MOTRA program in the MOLCAS program system.
The input for each module is preceded by its name like:
&MOTRA
8.32.3.1 Compulsory keywords
The following keywords are compulsory.| Keyword | Meaning |
| LUMOrb | Specifies that the molecular orbitals are read from a formatted file produced by one of the wave function generating programs. Note that either of Lumorb or Jobiph should be specified. LUMORB is the default keyword. No additional input is required. |
| JOBIph | Specifies that the molecular orbitals are read from a RASSCF job interface file. MOTRA will in this case read the average orbitals. No additional input is required. |
When natural orbitals from a RASSCF (or a state averaged CASSCF) calculation are to be used in MOTRA, they can be produced, or extracted from an existing JOBIPH file, by RASSCF, using keyword OUTOrbitals.
8.32.3.2 Optional keywords
There are a few useful optional keywords that can be specified. The following is a list
| Keyword | Meaning |
| AUTO | This keyword specified automatic deletion of orbitals based on occupation numbers. The following line contain one threshold per symmetry, and all orbitals with occupation numbers smaller that the threshold will be deleted. If AUTO and DELEte are both specified, the larger number will be used. |
| DELEted | Specifies the number of virtual orbitals that are not to be used as correlating orbitals in the subsequent CI calculation. The last orbitals in each symmetry are deleted. The default is no deleted orbitals. One additional line with the number of deleted orbitals in each symmetry (free format). |
| FROZen | Specifies the number of doubly occupied orbitals that are left uncorrelated in subsequent correlation calculation(s). Additional orbitals can be frozen in these programs, but from an efficiency point of view it is preferable to freeze orbitals in the transformation. One additional line with the number of frozen orbitals in each symmetry (free format). For more details on freezing orbitals in MOTRA see the program description. The frozen orbitals are the first in each symmetry block. Default is to freeze the core (but not semi-core) orbitals. |
| ONEL | Specifies that only one-electron integrals are to be transformed. No additional input is required. |
| Specifies the print level in the program. The default (1) does not print the orbitals that are used in the transformation, but they appear at print level 2. Beware of large print levels since vast amounts of output may be produced. The value is read from the line after the keyword, in free format. | |
| RFPErt | Add a constant reaction field perturbation to the bare nuclei Hamiltonian. The perturbation is read from the file RUNOLD (if not present defaults to RUNFILE) and is the latest self consistent perturbation generated by one of the programs SCF or RASSCF. |
| CTONly | Specifies that Cholesky vectors are to be transformed without subsequent calculation of the two-el integrals.
It requires as input one of the two following strings: "pqK" or "Kpq", which indicate the storage format as
L(pq,K) or L(K,pq), respectively. The former is the default option. Transformed vectors are stored in the files 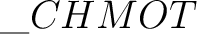 ,
one for each compound symmetry.
Available only in combination with Cholesky or RI integral representation. ,
one for each compound symmetry.
Available only in combination with Cholesky or RI integral representation.
|
| DIAGonal integrals | Activates the evaluation of the diagonal integrals in MO basis. Requires the keyword CTONly. The file DIAGINT is generated which contains these integrals. |
| TITLe | This keyword should be followed by exactly one title line. |
8.32.3.3 Input example
&MOTRA
Title = Water molecule.
* Don't correlate 1s on oxygen
Frozen = 1 0 0 0
Lumorb
Next: 8.33 mpprop Up: 8. Programs Previous: 8.31 mknemo

