
























MOLCAS manual: Next: 8.36 nemo Up: 8. Programs Previous: 8.34 MRCI
|
| File | Contents |
| UNSYM | Output file from the MCLR program |
8.35.2.2 Output files
| File | Contents |
| plot.intensity | Contains data for plotting an artificial spectrum.
|
8.35.3 Input
The input for MULA begins after the program name:
&MULA
There are no compulsory keyword.
8.35.3.1 Keywords
| Keyword | Meaning |
| TITLe | Followed by a single line, the title of the calculation. |
| FORCe | A force field will be given as input (or read from file), defining two oscillators for which individual vibrational levels and transition data will be computed. |
| ATOMs | Followed by one line for each individual atom in the molecule. On each line is the label of the atom, consisting of an element symbol followed by a number. After the label, separated by one or more blanks, one can optionally give a mass number; else, a standard mass taken from the file data/atomic.data. After these lines is one single line with the keyword "END of atoms". |
| INTErnal | Specification of which internal coordinates that are to be used in the calculation. Each subsequent line has the form 'BOND a b' or 'ANGLE a b c' or or 'TORSION a b c d' or or 'OUTOFPL a b c d', for bond distances, valence angles, torsions (e.g. dihedral angles), and out-of-plane angles. Here, a...d stand for atom labels. After these lines follows one line with the keyword "END of internal". |
| MODEs | Selection of modes to be used in the intensity calculation. This is followed by a list of numbers, enumerating the vibrational modes to use. The modes are numbered sequentially in order of vibrational frequency. After this list follows one line with the keyword "END of modes". |
| MXLEvels | Followed by one line with the maximum number of excitations in each of the two states. |
| VARIational | If this keyword is included, a variational calculation will be made, instead of using the default double harmonic approximation. |
| TRANsitions | Indicates the excitations to be printed in the output. Followed by the word FIRST on one line, then a list of numbers which are the number of phonons – the excitation level – to be distributed among the modes, defining the vibrational states of the first potential function (force field). Then similarly, after a line with the word SECOND, a list of excitation levels for the second state. |
| ENERgies | The electronic T0 energies of the two states, each value is followed by either "eV" or "au". |
| GEOMetry | Geometry input. Followed by keywords FILE, CARTESIAN, or INTERNAL. If FILE, the geometry input is taken from UNSYM1 and UNSYM2. If CARTESIAN or INTERNAL, two sections follow, one headed by a line with the word FIRST, the other with the word SECOND. For the CARTESIAN case, the following lines list the atoms and coordinates. On each line is an atom label, and the three coordinates (x,y,z). For the INTERNAL case, each line defines an internal coordinate in the same way as for keyword INTERNAL, and the value. |
| MXORder | Maximum order of transition dipole expansion. Next line is 0, if the transition dipole is constant, 1 if it is a linear function, etc. |
| OSCStr | If this keyword is included, the oscillator strength, instead of the intensity, of the transitions will calculated. |
| BROAdplot | Gives the peaks in the spectrum plot an artificial halfwidth. The default
lifetime is
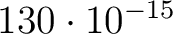 s but this can be changed with keyword
LIFEtime followd by the value. s but this can be changed with keyword
LIFEtime followd by the value.
|
| NANOmeters | If this keyword is included, the plot file will be in nanometers. Default is in eV. |
| CM-1 | If this keyword is included, the plot file will be in cm-1. Default is in eV. |
| PLOT | Enter the limits (in eV, cm-1, or in nm) for the plot file. |
| VIBWrite | If this keyword is included, the vibrational levels of the two states will be printed in the output. |
| VIBPlot | Two files, plot.modes1 and plot.modes2, will be generated, with pictures of the normal vibrational modes of the two electronic states. |
| HUGElog | This keyword will give a much more detailed output file. |
| SCALe | Scales the Hessians, by multiplying with the scale factors following this keyword. |
| DIPOles | Transition dipole data. If MXORDER=0 (see above), there follows a single line with x,y,z components of the transition dipole moment. If MXORDER=1 there are an additional line for each cartesian coordinate of each atom, with the derivative of the transition dipole moment w.r.t. that nuclear coordinate. |
| NONLinear | Specifies non-linear variable substitutions to be used in the definition of potential surfaces. |
| POLYnomial | Gives the different terms to be included in the fit of the polynomial to the energy data. |
| DATA | Potential energy surface data.
|
8.35.3.2 Input example
&MULA
Title
Water molecule
Atoms
O1
H2
H3
End Atoms
Internal Coordinates
Bond O1 H2
Bond O1 H3
Angle H3 O1 H2
End Internal Coordinates
MxLevels
0 3
Energies
First
0.0 eV
Second
3.78 eV
Geometry
Cartesian
First
O1 0.0000000000 0.0000000000 -0.5000000000
H2 1.6000000000 0.0000000000 1.1000000000
H3 -1.6000000000 0.0000000000 1.1000000000
End
Second
O1 0.0000000000 0.0000000000 -0.4500000000
H2 1.7000000000 0.0000000000 1.0000000000
H3 -1.7000000000 0.0000000000 1.0000000000
End
ForceField
First state
Internal
0.55 0.07 0.01
0.07 0.55 0.01
0.01 0.01 0.35
Second state
Internal
0.50 0.03 0.01
0.03 0.50 0.01
0.01 0.01 0.25
DIPOles
0.20 0.20 1.20
BroadPlot
LifeTime
10.0E-15
NANO
PlotWindow
260 305
End of input
&MULA
TITLe
Benzene
ATOMs
C1
C2
C3
C4
C5
C6
H1
H2
H3
H4
H5
H6
End of Atoms
GEOMetry
file
INTERNAL COORDINATES
Bond C1 C3
Bond C3 C5
Bond C5 C2
Bond C2 C6
Bond C6 C4
Bond C1 H1
Bond C2 H2
Bond C3 H3
Bond C4 H4
Bond C5 H5
Bond C6 H6
Angle C1 C3 C5
Angle C3 C5 C2
Angle C5 C2 C6
Angle C2 C6 C4
Angle H1 C1 C4
Angle H2 C2 C5
Angle H3 C3 C1
Angle H4 C4 C6
Angle H5 C5 C3
Angle H6 C6 C2
Torsion C1 C3 C5 C2
Torsion C3 C5 C2 C6
Torsion C5 C2 C6 C4
Torsion H1 C1 C4 C6
Torsion H2 C2 C5 C3
Torsion H3 C3 C1 C4
Torsion H4 C4 C6 C2
Torsion H5 C5 C3 C1
Torsion H6 C6 C2 C5
END INTERNAL COORDINATES
VIBPLOT
cyclic 4 1
ENERGIES
First
0.0 eV
Second
4.51 eV
MODES
14 30 5 6 26 27 22 23 16 17 1 2 9 10
END
MXLE - MAXIMUM LEVEL of excitation (ground state - excited state)
2 2
MXOR - MAXIMUM ORDER in transition dipole.
1
OscStr
Transitions
First
0
Second
0 1 2
FORCEFIELD
First
file
Second
file
DIPOLES
file
Next: 8.36 nemo Up: 8. Programs Previous: 8.34 MRCI

