
























MOLCAS manual: Next: 8.46 single_aniso Up: 8. Programs Previous: 8.44 scf
The SEWARD module generates one- and two-electron integrals needed
by other programs. The two-electron integrals may optionally be
Cholesky decomposed. In addition, it will serve as the input parser
for parameters related to the specification of the quadrature grid
used in numerical integration in association with DFT and reaction
field calculations.
|
| Max number of unique basis functions: | 2000 |
| Max number of symmetry independent centers: | 500 |
| Highest angular momentum: | 15 |
| Highest symmetry point group: | D2h |
8.45.1.2 Dependencies
SEWARD usually runs after program GATEWAY. In the same time, any input used
in GATEWAY can be placed into SEWARD input. However, it is recommended to
specify all information about the molecule and the basis set in GATEWAY input.
SEWARD does normally not depend on any other code, except of GATEWAY. There are two exceptions however. The first one is when SEWARD is used as a property module. In these cases the file INPORB has to have been generated by a wave function code. The second case, which is totally transparent to the user, is when SEWARD picks up the new Cartesian coordinates generated by SLAPAF during a geometry optimization.
8.45.1.3 Files
8.45.1.3.1 Input Files
Apart form the standard input file
SEWARD will use the following input files: RYSRW, ABDATA,
RUNFILE, INPORB (for calculation of properties) (![[*]](crossref.png) ).
In addition, SEWARD uses the following files:
).
In addition, SEWARD uses the following files:
| File | Contents |
| BASLIB | The default directory for one-particle basis set information.
This directory contains files which are part
of the program system and could
be manipulated by the user in accordance with the instructions in the
section and following subsections.
New basis set files can be added to this directory by the local
MOLCAS administrator.
|
| QRPLIB | Library for numerical mass-velocity plus Darwin potentials (used for ECPs). |
8.45.1.3.2 Output files
In addition to the standard output file
SEWARD may generate the following files:
ONEINT, ORDINT, CHVEC, CHRED, CHORST,
CHOMAP, CHOR2f ().
8.45.1.4 Input
Below follows a description of the input to SEWARD.
Observe that if
nothing else is requested
SEWARD will by default compute the overlap, the
dipole, the quadrupole, the nuclear attraction, the kinetic energy,
the one-electron Hamiltonian, and the two-electron repulsion
integrals.
The input for each module is preceded by its name like:
&SEWARD
Argument(s) to a keyword, either individual or composed by several entries, can be placed in a separated line or in the same line separated by a semicolon. If in the same line, the first argument requires an equal sign after the name of the keyword.
8.45.1.4.1 General keywords
| Keyword | Meaning | ||||||||||
| TITLe | One line of title card follows. | ||||||||||
| TEST | SEWARD will only process the input and generate a non-zero return code. | ||||||||||
| ONEOnly | SEWARD will not compute the two-electron integrals. | ||||||||||
| NODKroll | SEWARD will not compute Douglas-Kroll integrals. | ||||||||||
| DIREct | Prepares for later integral-direct calculations. As with keyword OneOnly, SEWARD will evaluate no two-electron integrals. | ||||||||||
| EXPErt | Sets ``expert mode'', in which various default settings are altered. Integral-direct calculations will be carried out if the two-electron integral file is unavailable. | ||||||||||
| CHOLesky | SEWARD will Cholesky decompose the two-electron integrals using default configuration (in particular, the decomposition threshold is 1.0d-4) of the decomposition driver. The decomposition threshold can be changed using keyword THRC. Default is to not decompose. | ||||||||||
| 1CCD | SEWARD will Cholesky decompose the two-electron integrals using the one-center approximation. The decomposition threshold can be changed using keyword THRC. Default is to not decompose. | ||||||||||
| THRCholesky | Specify decomposition threshold for Cholesky decomposition of two-electron integrals on the next line. Default value: 1.0d-4. | ||||||||||
| SPAN | Specify span factor (
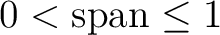 ) for Cholesky decomposition of two-electron integrals on the next line.
Span=1 implies full pivoting, Span=0 means no pivoting. If the span factor is too low, numerical errors may cause
negative diagonal elements and force the program to quit; if the span factor is too large, the execution time may
increase. Default value: 1.0d-2. ) for Cholesky decomposition of two-electron integrals on the next line.
Span=1 implies full pivoting, Span=0 means no pivoting. If the span factor is too low, numerical errors may cause
negative diagonal elements and force the program to quit; if the span factor is too large, the execution time may
increase. Default value: 1.0d-2.
| ||||||||||
| LOW Cholesky | SEWARD will Cholesky decompose the two-electron integrals using low accuracy (threshold 1.0d-4) configuration of the decomposition driver. Default is to not decompose. | ||||||||||
| MEDIum Cholesky | SEWARD will Cholesky decompose the two-electron integrals using medium accuracy (threshold 1.0d-6) configuration of the decomposition driver. Default is to not decompose. | ||||||||||
| HIGH Cholesky | SEWARD will Cholesky decompose the two-electron integrals using high-accuracy (threshold 1.0d-8) configuration of the decomposition driver. Default is to not decompose. | ||||||||||
| LDF | Local Density Fitting: SEWARD will fit each AO product using only auxiliary functions centered on the two parent atoms. Fitting coefficients are computed and stored for use in later modules (only non-hybrid KS-DFT at the moment). Requires an auxiliary basis set (generated by RICD in Gateway or externally defined). Can not be combined with Cholesky and (global) DF/RI. LDF is turned off by default. | ||||||||||
| LDF1 | Local Density Fitting using auxiliary functions centered on the two parent atoms of each AO product. Equivalent to keyword LDF. Equivalent to keyword LDF. | ||||||||||
| LDF2 | LDF using auxiliary functions centered on the two parent atoms of each AO product as well as the two-center functions required to achieve the given target accuracy. | ||||||||||
| TARGet accuracy | Specify the target accuracy for LDF2 on the next line. Default value: threshold used to generate the aCD or acCD auxiliary basis set (or 1.0d-4 in case of externally defined auxiliary basis sets). | ||||||||||
| APTH | Specify the LDF/LDF2 atom pair prescreening threshold on the next line, thus defining which atom pairs are considered significant. Default value: the target accuracy or the cutoff threshold for primitive integral evaluation, whichever is smaller. | ||||||||||
| CLDF | Constrained LDF/LDF2: specify constraint order on the next line (-1 for unconstrained, 0 for charge constraint). Default: unconstrained LDF. | ||||||||||
| FAKE CD/RI | If CD/RI vectors are already available, SEWARD will not redo work! | ||||||||||
| JMAX | The integer entry on the next line is the highest rotational quantum number for which SEWARD will compute the rotational energy within the rigid rotor model. The default value is 5. | ||||||||||
| SYMMetry | See the the description in the manual for the program GATEWAY | ||||||||||
| BASIs Set | See the the description in the manual for the program GATEWAY | ||||||||||
| ZMAT | See the the description in the manual for the program GATEWAY | ||||||||||
| XBAS | See the the description in the manual for the program GATEWAY | ||||||||||
| XYZ | See the the description in the manual for the program GATEWAY | ||||||||||
| NOGUessorb | Disable automatic generation of starting orbitals with the GuessOrb procedure. | ||||||||||
| NODElete | Do not delete any orbitals automatically. | ||||||||||
| SDELete | Set the threshold for deleting orbitals based on the eigenvalues of the overlap matrix. All eigenvalues with eigenvectors below this threshold will be deleted. If you want no orbitals deleted use keyword NODElete. | ||||||||||
| TDELete | Set the threshold for deleting orbitals based on the eigenvalues of the kinetic energy matrix. All eigenvalues with eigenvectors above this threshold will be deleted. If you want no orbitals deleted use keyword NODElete. | ||||||||||
| ECPShow | Force SEWARD to print ECP parameters. | ||||||||||
| AUXShow | Force SEWARD to print auxiliary basis set parameters. | ||||||||||
| BSSHow | Force SEWARD to print basis set parameters. | ||||||||||
| VERBose | Force SEWARD to print a bit more verbose. | ||||||||||
| EMBEdding | Reads in an embedding potential from a file. It can also write out the
density and the electrostatic potential on a grid. It is a block keyword
which MUST be ended with ENDEmbedding.
The subkeywords are:
|
8.45.1.4.2 Keywords associated to one-electron integrals
| Keyword | Meaning |
| MULTipoles | Entry which specifies the highest order of the multipole for which integrals will be generated. The default center for the dipole moment operator is the origin. The default center for the higher order operators is the center of the nuclear mass. The default is to do up to quadrupole moment integrals (2). |
| CENTer | This option is used to override the default selection of the origin of the multipole moment operators. On the first entry add an integer entry specifying the number of multipole moment operators for which the origin of expansion will be defined. Following this, one entry for each operator, the order of the multipole operator and the coordinates of the center (in au) of expansion are specified. |
| SDIPole | Requests computation of velocity integrals. |
| RELInt | Requests the computation of mass-velocity and one-electron Darwin contact term integrals for the calculation of a first order correction of the energy with respect to relativistic effects. |
| RXXPyy | Request arbitrary scalar relativistic Douglas-Kroll-Hess (DKH) correction to the one-electron Hamiltonian
and the so-called picture-change correction to the property integrals (multipole moments
and electronic potential related properties).
Here XX represents the order of the DKH correction to the one-electron Hamiltonian and
yy the order of the picture-change correction. The character P denotes the parameterization
used in the DKH procedure.
The possible parametrizations P of the unitary transformation used
in the DKH transformation supported by MOLCAS are:
|
| RX2C | Request the scalar relativistic X2C (eXact-two-Component) corrections to the one-electron Hamiltonian as well as the property integrals. |
| RBSS | Request the scalar relativistic BSS (Barysz-Sadlej-Snijders) corrections to the one-electron Hamiltonian as well as the property integrals. The non-iterative scheme is employed for the construction of BSS transformation. |
| RLOCal | Request local approximation to the relativistic exact decoupling approaches such as X2C, BSS and DKH. This option cannot be used together with point group symmetry. |
| NOAMfi | Explicit request for no computation of atomic mean-field integrals. |
| AMFI | Explicit request for the computation of atomic mean-field integrals (used in subsequent spin-orbit calculations). These integrals are computed by default for the ANO-RCC and ANO-DK3 basis sets. |
| Grid Input | Specification of numerical quadrature parameters, consult the numerical quadrature section of this manual. |
8.45.1.4.3 Additional keywords for property calculations
| Keyword | Meaning |
| VECTors | Requests a property calculation. For this purpose a file, INPORB, must be available, which contains the MO's and occupation numbers of a wave function. A custom filename can be given with FileOrb. |
| FILEorb | The next line specifies the filename containing the orbitals and occupation numbers from which the properties will be computed. By default a file named INPORB will be used. |
| ORBCon | The keyword will force SEWARD to produce a list of the orbital contributions to the properties being computed. The default is to generate a compact list. |
| THRS | The real entry on the following line specifies the threshold for the occupation number of an orbital in order for the OrbCon option to list the contribution of that orbital to a property. The default is 1.0d-6. |
| ORBAll | When OrbCon is present, the keyword will force SEWARD to produce a list of the computed properties of all orbitals (including unoccupied orbitals), and the properties are not weighted by orbital occupation numbers. The total electronic and nuclear contributions printed are the same as those printed by using OrbCon without OrbAll. |
8.45.1.4.4 Keywords for two-electron integrals
| Keyword | Meaning |
| NOPAck | The two-electron integrals will not be packed. The default is to pack the two-electron integrals. |
| PKTHre | An entry specifies the desired accuracy for the packing algorithm, the default is 1.0d-14. |
| STDOut | Generate a two-electron integral file according to the standard of version 1 of MOLCAS. The default is to generate the two-electron integrals according to the standard used since version 2 of MOLCAS. |
| THREshold | Threshold for writing integrals to disk follows. The default is 1.0d-14. |
| CUTOff | Threshold for ignoring the calculation of integrals based on the pair prefactor follows. The default is 1.0d-16. |
8.45.1.4.5 Keywords associated to electron-molecule scattering calculations within the framework of the R-matrix method
This section contains keyword which control the radial numerical integration of the diffuse basis functions describing the scattered electrons in the variational R-matrix approach. The activation of this option is controlled by that the center of the diffuse basis is assigned the unique atom label DBAS.
| Keyword | Meaning |
| RMAT | Radius of the R-matrix sphere (in Bohr). This sphere is centered at the coordinate origin. The default is 10 Bohr. |
| RMEA | Absolute precision in radial integration. The default is 1d-9. |
| RMER | Relative precision in radial integration. The default is 1d-14. |
| RMQC | Effective charge of the target molecule. This is the effective charge seen by the incident electron outside of the R-matrix sphere. The default is 0d0. |
| RMDI | Effective dipole of the target molecule. This is the effective dipole seen by the incident electron outside of the R-matrix sphere. The default is (0d0,0d0,0d0). |
| RMEQ | Minimal value of the effective charge of the target molecule to be considered. This is also the minimal value of the components of the effective dipole to be considered. Default is 1d-8 |
| RMBP | Parameter used for test purposes in the definition of the Bloch term. Default is 0d0. |
| CELL | Defines the three vectors of the unit cell (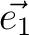 , ,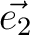 , ,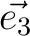 ).
The optional keyword
Angstrom before the definition of vectors would read data in Å.
Must consist of three entries (four in the case of Å) which correspond to coordinates of the vectors. All the atoms which
are defined after that key are considered as the atoms of the cell. ).
The optional keyword
Angstrom before the definition of vectors would read data in Å.
Must consist of three entries (four in the case of Å) which correspond to coordinates of the vectors. All the atoms which
are defined after that key are considered as the atoms of the cell.
|
| SPREad | Three integer numbers n1, n2, n3 which define the spread of the unit cell along the unit cell vectors.
For example,
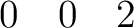 would add all cell's atoms translated on would add all cell's atoms translated on 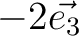 , , 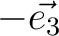 , , , , 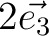 .
This key must be placed before the definition of the unit cell atoms. .
This key must be placed before the definition of the unit cell atoms.
|
Below follows an input for the calculation of integrals of a carbon atom. The comments in the input gives a brief explanation of the subsequent keywords.
&SEWARD
Title= This is a test deck!
* Remove integrals from a specific irreps
Skip= 0 0 0 0 1 1 1 1
* Requesting only overlap integrals.
Multipole= 0
* Request integrals for diamagnetic shielding
DSHD= 0.0 0.0 0.0; 1; 0.0 0.0 0.0
* Specify a title card
* Request only one-electron integrals to be computed
OneOnly
* Specify group generators
Symmetry= X Y Z
* Enable an inline basis set
Expert
* Specify basis sets
Basis set
C.ANO-L...6s5p3d2f.
Contaminant d
C 0.0 0.0 0.0
End of basis
8.45.1.4.6 The basis set label and the all electron basis set library
The label, which defines the basis set for a given atom or set of atoms, is given as input after the keyword Basis set. It has the following general structure (notice that the last character is a period):

where the different identifiers have the following meaning:
| Identifier | Meaning |
| atom | Specification of the atom by its chemical symbol. |
| type | Gives the type of basis set (ANO, STO, ECP, etc.) according to specifications given in the basis set library, vide supra. Observe that the upper cased character of the type label defines the file name in the basis directory. |
| author | First author in the publication where that basis set appeared. |
| primitive | Specification of the primitive set (e.g. 14s9p4d3f). |
| contracted | Specification of the contracted set to be selected. Some basis sets allow only one type of contraction, others all types up to a maximum. The first basis functions for each angular momentum is then used. Note, for the basis set types ANO and ECP, on-the-fly decontraction of the most diffuse functions are performed in case the number of contracted functions specified in this field exceeds what formally is specified in the library file. |
| aux | Specification of the type of AIMP, for instance, to choose between non-relativistic and relativistic core AIMP's. |
atom, type,
and contracted have to be included in
the label. The others can be left out. However, the periods have to be
kept. Example — the basis set label
`C.ano-s...4s3p2d.'
will by MOLCAS be
interpreted as
`C.ano-s.Pierloot.10s6p3d.4s3p2d.',
which is the first
basis set in the ANO-S file in the
basis directory that fulfills the specifications given.
More information about basis set format can be found in the section Advanced examples.
8.45.2 Numerical integration
Various Density Functional Theory (DFT) models can be used in MOLCAS . Energies and analytical gradients are available for all DFT models. In DFT the exact exchange present in HF theory is replaced by a more general expression, the exchange-correlation functional, which accounts for both the exchange energy, EX [P] and the electron correlation energy ,EC [P].
8.45.2.1 Description
We shall now describe briefly how the exchange and correlation energy terms look like.
The functionals used in DFT are integrals of some function of the electron density and optionally the gradient
of the electron density
![\begin{displaymath}
E_X[P] = \int f(\rho_{\alpha}(r), \rho_{\beta}(r), \nabla \rho_{\alpha}(r), \nabla \rho_{\beta}(r)) dr
\end{displaymath}](img182.png) |
(8.18) |
The various DFT methods differ in which function, f, is used for EX[P] and for EC[P]. In MOLCAS pure DFT methods are supported, together with hybrid methods, in which the exchange functional is a linear combination of the HF exchange and a functional integral of the above form. The latter are evaluated by numerical quadrature. In the SEWARD input the parameters for the numerical integration can be set up. In the SCF and RASSCF inputs the keywords for using different functionals can be specified. Names for the various pure DFT models are given by combining the names for the exchange and correlation functionals.
The DFT gradients has been implemented for both the fixed and the moving grid approach [128,129,130]. The latter is known to be translationally invariant by definition and is recommended in geometry optimizations.
8.45.2.2 Input
Below follows a description of the input to the numerical integration utility in the SEWARD input.
Compulsory keywords
| Keyword | Meaning |
| GRID Input | This marks the beginning of the input to the numerical integration utility. |
| END Of Grid-Input | This marks the end of the input to the numerical integration utility. |
Optional keywords
| Keyword | Meaning |
| GRID | It specifies the quadrature quality. The possible indexes that can follow are COARSE, SG1GRID, FINE, ULTRAFINE following the Gaussian98 convention. Default is FINE. |
| RQUAd | It specifies the radial quadrature scheme. Options are LOG3 (Mura and Knowles)[131], BECKE (Becke)[132], MHL (Murray et al.)[133], TA (Treutler and Ahlrichs, defined for H-Kr)[134], and LMG (Lindh et al.)[135], respectively. The default is MHL. |
| GGL | It activates the use of Gauss and Gauss-Legendre angular quadrature. Default is to use the Lebedev angular grid. |
| LEBEdev | It turns on the Lebedev angular grid. |
| LOBAtto | It activates the use of Lobatto angular quadrature. Default is to use the Lebedev angular grid. |
| LMAX | It specifies the angular grid size. Default is 29. |
| NGRId | It specifies the maximum number of grid points to process at one instance. Default is 128 grid points. |
| NOPRunning | It turns off the the angular prunning. Default is to prune. |
| NR | It is followed by the number of radial grid points. Default is 75 radial grid points. |
| FIXEd grid | Use a fixed grid in the evaluation of the gradient. This corresponds to using the grid to numerically evaluate the analytic gradient expression. Default is to use a moving grid. |
| MOVIng grid | Use a moving grid in the evaluation of the gradient. This correspond to evaluating the gradient of the numerical expression of the DFT energy. This is the default. |
| THREshold | It is followed by the value for the the radial threshold. Default value is 1.0D-13. |
| T_X | Threshold for screening in the assembling of the density on the grid. Default value is 1.0D-18. |
| T_Y | Threshold for screening in the assembling of the integrals. Default value is 1.0D-11. |
| NOSCreening | Turn off any screening in the numerical integration. |
| CROWding | The crowding factor, according to MHL, used in the pruning of the angular grid close to the nuclei. Default value 3.0. |
The SCF and RASSCF programs have their own keywords to decide which functionals to use in a DFT calculation.
Below follows an example of a DFT calculation with two different functionals.
&GATEWAY
Basis set
H.3-21G.....
H1 0.0 0.0 0.0
End of basis
&SEWARD
Grid input
RQuad= Log3; nGrid= 50000; GGL; lMax= 26; Global
End of Grid Input
&SCF; Occupations=1; KSDFT=LDA5; Iterations= 1 1
&SCF; Occupations= 1; KSDFT=B3LYP; Iterations= 1 1
8.45.3 Relativistic operators
The current different implementation of all relativistic operators in MOLCAS as described in the following subsubsections has been programmed and tested in Ref.[136]
8.45.3.1 Using the Douglas–Kroll–Hess Hamiltonian
For all-electron calculations, the preferred way is to use the scalar-relativistic Douglas–Kroll–Hess (DKH) Hamiltonian, which, in principle, is available up to arbitrary order in Molcas; for actual calculations, however, the standard 2nd order is usually fine, but one may use a higher order that 8th order by default to be on the safe side.
The arbitrary-order Hamiltonian is activated by setting
RXXPyy
somewhere in the SEWARD input, where the XX denotes the order of the DKH Hamiltonian in the external potential. I.e., for the standard 2nd-order Hamiltonian you may use R02O. Note in particular that the parametrization P does not affect the Hamiltonian up to fourth order. Therefore, as long as you run calculations with DKH Hamiltonians below 5th order you may use any symbol for the parametrization as they would all yield the same results.
The possible parametrizations P of the unitary transformation used in the DKH transformation supported by MOLCAS are:
- P=O:
- Optimum parametrization (OPT)
- P=E:
- Exponential parametrization (EXP)
- P=S:
- Square-root parametrization (SQR)
- P=M:
- McWeeny parametrization (MCW)
- P=C:
- Cayley parametrization (CAY)
Up to fourth order (XX=04) the DKH Hamiltonian is independent of the chosen parametrization. Higher-order DKH Hamiltonians depend slightly on the chosen parametrization of the unitary transformations applied in order to decouple the Dirac Hamiltonian. Since the EXP parameterization employs a fast algorithm [137], it is recommended for high-order DKH transformation.
For details on the arbitrary-order DKH Hamiltonians see [138] with respect to theory, [139] with respect to aspects of implementation, and [140] with respect to general principles of DKH. The current version of MOLCAS employs different algorithms, but the polynomial cost scheme of the DKH implementation as described in [137] is used as the default algorithm. The implementation in MOLCAS has been presented in [136].
For details on the different parametrizations of the unitary transformations see [141].
8.45.3.2 Douglas–Kroll–Hess transformed properties
As mentioned above, four-component molecular property operators need to be DKH transformed as well when going from a four-component to a two- or one-component description; the results do not coincide with the well-known corresponding nonrelativistic expressions for a given property but are properly picture change corrected.
The transformation of electric-field-like molecular property operators can be carried out for any order smaller or equal to the order chosen for the scalar-relativistic DKH Hamiltonian. In order to change the default transformation of order 2, you may concatenate the input for the DKH Hamiltonian by 2 more numbers specifying the order in the property,
RxxPyy
where yy denotes the order of the Hamiltonian starting with first order 01. The DKH transformation is then done automatically for all one-electron electric-field-like one-electron property matrices.
Also note that the current implementation of both the Hamiltonian and the property operators is carried out in the full, completely decontracted basis set of the molecule under consideration. The local nature of the relativistic contributions is not yet exploited and hence large molecules may require considerable computing time for all higher-order DKH transformations.
For details on the arbitrary-order DKH properties see [142] with respect to theory and [143,136] with respect to implementation aspects.
8.45.3.3 Using the X2C/Barysz–Sadlej–Snijders Hamiltonian
Exact decoupling of the relativistic Dirac Hamiltonian can be achieved with infinite-order approaches, such as the so-called X2C (exact–two–component) and BSS (Barysz–Sadlej–Snijders) method. In Molcas, both methods are available for all-electron calculations. The evaluation of transformation matrices employs a non-iterative scheme.
The exact decoupling Hamiltonian is activated by setting either RX2C or RBSS somewhere in the SEWARD input, where RX2C and RBSS denote using the scalar (one-component) X2C and BSS Hamiltonian respectively. The one-electron Hamiltonian as well as the property integrals will be transformed according to the given exact decoupling method. In other words, all property integrals are by default picture change corrected.
The computation time of the X2C/BSS method is almost the same as of the DKH method at 8th order, while X2C is a little bit faster than BSS since the additional free-particle Foldy–Wouthuysen transformation is skipped in the X2C approach[136]. For molecules including only light atoms, the DKH method with low orders (<8) is enough to account for the relativistic effects.
The differences between different exact decoupling relativistic methods are very small compared to errors introduced by other approximations, such as the basis set incompleteness, approximate density functionals, etc. Therefore, any exact decoupling model is acceptable for the treatment of relativistic effects in molecular calculations.
For details on the exact decoupling approaches see [136] with respect to theories and comparison of numerical results, [144,145,146] for the X2C method, and [147,148] for the BSS method.
8.45.3.4 Local approximation to relativistic decoupling
The computational cost for relativistic transformations increases rapidly if the molecule becomes larger. The local DLU scheme [149] was proposed to reduce the computational cost based on the atomic decomposition of the (unitary) decoupling transformation. It is important to note that the DLU scheme can be applied to any kind of relativistic approaches mentioned above (i.e., DKH, BSS, and X2C). It was found [149] that the DLU approximation introduces very small errors for total energies, which are less than 0.01 milli-Hartree for molecules including heavy atoms. The local approximation is activated by setting RLOCal somewhere in the SEWARD input.
The direct local approximation to the Hamiltonian, called DLH in Ref. [149] may be activated by setting RLOCal=DLH. However, as DLH is not superior to the DLU scheme, but introduces slightly larger errors, it is not recommended.
The picture-change effect for molecular properties is automatically taken care of when a local approximation is employed for the transformed operator. The default order of DKH transformed properties is set to the same as the order of the DKH Hamiltonian.
It is important to note the local approximation cannot be used together with point group symmetry in the current implementation. Because the relativistic transformation is applied in the molecular orbital (MO) representation instead of the atomic orbital (AO) representation. Thus, the program will report an error and exit if symmetry is used.
Next: 8.46 single_aniso Up: 8. Programs Previous: 8.44 scf

