
























MOLCAS manual: Next: 5.2 Optimizing geometries Up: 5. Problem Based Tutorials Previous: 5. Problem Based Tutorials
5.1 Electronic Energy at Fixed Nuclear GeometryThe MOLCAS 8.1 suite of Quantum Chemical programs is modular in design, and a desired calculation is achieved by executing a list of MOLCAS program modules in succession, occasionally manipulating the program information files. If the information files from a previous calculation are saved, then a subsequent calculation need not recompute them. This is dependent on the correct information being preserved in the information files for the subsequent calculations. Each module has keywords to specify the functions to be carried out, and many modules rely on the specification of keywords in previous modules. In the present examples the calculations will be designed by preparing a single file in which the input for the different programs is presented sequentially. The initial problem will be to compute an electronic energy at a fixed geometry of the nuclei, and this will be performed using different methods and thus requiring different MOLCAS program modules. First, the proper MOLCAS environment has to be set up which requires that following variables must be properly defined, for instance:
If not defined, MOLCAS provides default values for the above environment variables:
The first run involves a calculation of the SCF energy of the methane (CH4) molecule. Three programs should be used: GATEWAY to specify information about the system, SEWARD to compute and store the one- and two-electron integrals, and SCF to obtain the Hartree-Fock SCF wave function and energy.
The three MOLCAS programs to
be used leads to three major entries in the input file: GATEWAY, SEWARD, and SCF.
The GATEWAY program contains the nuclear geometry in cartesian
coordinates and the label for the one-electron basis set.
The keyword coord allows automatic insertion of GATEWAY input from a standard
file containing the cartesian coordinates in Angstrom which can be generated by
programs like LUSCUS or MOLDEN).
No symmetry is being considered so the keyword group=C1 is used to force the program not
to look for symmetry in the CH4 molecule, and ,thus, input for SEWARD is not required.
In closed-shell cases, like CH4, input for SCF is not required. All the input
files discussed here can be found at
where the content of the CH4.xyz file is:
To run MOLCAS , simply execute the command
or
The most relevant information is contained in the output file, where the GATEWAY program information describing the nuclear geometry, molecular symmetry, and the data regarding the one-electron basis sets and the calculation of one- and two-electron integrals, as described in section 6.4. Next, comes the output of program SCF with information of the electronic energy, wave function, and the Hartree-Fock (HF) molecular orbitals (see section 6.5). Files containing intermediate information, integrals, orbitals, etc, will be kept in the $WorkDir directory for further use. For instance, files $Project.OneInt and $Project.OrdInt contain the one- and two-electron integrals stored in binary format. File $Project.ScfOrb stores the HF molecular orbitals in ASCII format, and $Project.RunFile is a communication file between programs. All these files can be used later for more advanced calculations avoiding a repeat of certain calculations. There are graphical utilities that can be used for the analysis of the results. By default, MOLCAS generates files which can be read with the MOLDEN program and are found in the $WorkDir including the fileCH4.scf.molden. This file contains information about molecular geometry and molecular orbitals, and requires the use if Density Mode in MOLDEN. However, MOLCAS has its own graphical tool, program LUSCUS, which is a viewer based on openGL and allows the visualization of molecular geometries, orbitals, densities, and density differences. For example, a graphical display of the CH4 molecule can be obtained from a standard coordinate file by the following command:
In order to obtain the information for displaying molecular orbitals and densities, it is necessary to run the MOLCAS program called GRID_IT:
Now, execcute the MOLCAS program:
In the $WorkDir and $PWD directories a new file is generated, CH4.lus which contains the information required by the GRID_IT input. The file can be visualized by LUSCUS (Open source program, which can be downloaded and installed to your Linux, Windows, or MacOS workstation or laptop). By typing the command:
a window will be opened displaying the molecule and its charge density. By proper selection of options with the mouse buttons, the shape and size of several molecular orbitals can be visualized. GRID_IT can also be run separately, if an orbital file is specified in the input, and the $WorkDir directory is available.
More information can be found in section As an alternative to running a specific project, the short script provided below can be placed in the directory $MOLCAS/doc/samples/problem_based_tutorials with the name project.sh. Simply execute the shell script, project.sh $Project, where $Project is the MOLCAS input, and output files, error files, and a $WorkDir directory called $Project.work will be obtained.
In order to run a Kohn-Sham density functional calculation, MOLCAS uses the same SCF module, and, therefore, the only change needed are the specification of the DFT option and required functional (e.g. B3LYP) in the SCF input:
Similar graphical files can be found in $WorkDir and $PWD. The next step is to obtain the second-order Møller–Plesset perturbation (MP2) energy for methane at the same molecular geometry using the same one-electron basis set. Program MBPT2 is now used, and it is possible to take advantage of having previously computed the proper integrals with SEWARD and the reference closed-shell HF wave function with the SCF program. In such cases, it is possible to keep the same definitions as before and simply prepare a file containing the MBPT2 input and run it using the molcas command. The proper intermediate file will be already in $WorkDir. On the other hand, one has to start from scratch, all required inputs should be placed sequentially in the MP2.energy.CH4 file. If the decision is to start the project from the beginning, it is probably a good idea to remove the entire $WorkDir directory, unless it is known for certain the exact nature of the files contained in this directory.
In addition to the calculation of a HF wave function, an MP2 calculation has been performed with a frozen deepest orbital, the carbon 1s, of CH4. Information about the output of the MBPT2 program can be found on section 6.6. The SCF program works by default with closed-shell systems with an even number of electrons at the Restricted Hartee-Fock (RHF) level. If, instead there is a need to use the Unrestricted Hartree Fock (UHF) method, this can be schieved by invoking the keyword UHF. This is possible for both even and odd electron systems. For instance, in a system with an odd number of electrons such as the CH3 radical, with the following Cartesian coordinates
the input to run an open-shell UHF calculation is easily obtained:
If the system is charged, this must be indicated in the SCF input, for example, by computing the cation of the CH4 molecule at the UHF level:
The Kohn-Sham DFT calculation can be also run using the UHF algorithm:
For the UHF and UHF/DFT methods it is also possible to specify
Different sets of methods use other MOLCAS modules. For example, to perform a Complete Active Space (CAS) SCF calculation, the RASSCF program has to be used. This module requires starting trial orbitals, which can be obtained from a previous SCF calculation or, automatically, from the SEWARD program which provides trial orbitals by using a model Fock operator. Recommended keywords are
In this case, the lowest singlet state (i.e. the ground dstate) is computed, since this is a closed-shell situation with an active space of eight electrons in eight orbitals and with an inactive C 1s orbital, the lowest orbital of the CH4 molecule. This is a CASSCF example in which all the valence orbitals and electrons (C 2s, C 2p and 4 x H 1s) are included in the active space and allows complete dissociation into atoms. If this is not the goal, then the three almost degenerate highest energy occupied orbitals and the corresponding antibonding unoccupied orbitalsmust be active, leading to a 6 in 6 active space. Using the CASSCF wave function as a reference, it is possible to perform a second-order perturbative, CASPT2, correction to the electronic energy by employing the CASPT2 program. If all previously calculated files are retained in the $WorkDir directory, in particular, integral files (CH4.OneInt,CH4.OrdInt), the CASSCF wave function information file (CH4.JobIph), and communication file CH4.RunFile), it will not be necessary to re-run programs SEWARD, and RASSCF. In this case case, it is enough to prepare a file containing input only for the CASPT2 program followed be execution. Here, however, for the sake of completness, input to all MOLCAS moddules is provided:
In most of casesi, the Hartree-Fock orbitals will be a better choice as starting orbitals. In that case, the RASSCF input has to include keyword LumOrb to read from any external source of orbitals other than those generated by the SEWARD program. By modifying input to the SCF program, it is possible to generate alternative trial orbitals for the RASSCF program. Since a new set of trial orbitals is used, the input to the RASSCF program is also changed. Now, the number of active orbitals, as well as the number of active electrons, are 6. The two lowest orbitals (Inactive 2) are excluded from the active space and one other orbital is placed in the secondary space. If the previous (8,8) full valence space was used, the CASPT2 program would not be able to include more electronic correlation energy, considering that the calculation involves a minimal basis set. The input for the CASPT2 program includes a frozen C 1s orbital, the lowest orbital in the CH4 molecule. The charge and multiplicity of our wave function can be changed by computing the CH4+ cation with the same methods. The RASSCF program defines the character of the problem by specifying the number of electrons, the spin multiplicity, and the spatial symmetry. In the example below, there is one less electron giving rise to doublet multiplicity:
No further modification is needed to the CASPT2 input:
A somewhat more sophisticated calculation can be performed at the Restricted Active Space (RAS) SCF level. In such a situation, the level of excitation in the CI expansion can be controlled by restricting the number of holes and particles present in certain orbitals.
In particular, the previous calculation includes one orbital within the Ras1 space and one orbital within the Ras3 space. One hole (single excitation) at maximum is allowed from Ras1 to Ras2 or Ras3, while a maximum of one particle is allowed in Ras3, derived from either Ras1 or Ras2. Within Ras2, all types of orbital occupations are allowed. The RASSCF wave functions can be used as reference for multiconfigurational perturbation theory (RASPT2), but this approach has not been as extensively tested as CASPT2, and, so experience is still somewhat limited. MOLCAS also has the possibility of computing electronic energies at different CI levels by using the MRCI program. The input provided below involves a Singles and Doubles Configuration Interaction (SDCI) calculation on the CH4 molecule. To set up the calculations, program MOTRA which transforms the integrals to molecular basis, and program GUGA which computes the coupling coefficients, must be run before the MRCI program. In MOTRA the reference orbitals are specifiedi, and those employed here are from an HF SCF calculation including frozen orbitals. In GUGA the reference for the CI calculation is described by the number of correlated electrons, the spatial and spin symmetry, the inactive orbitals always occupation 2 in the reference space, and the type of CI expansion.
To use reference orbitals from a previous CASSCF calculation, the RASSCF program will have to be run before the MOTRA module. Also, if the spatial or spin symmetry are changed for the CI calculation, the modifications will be introduced in the input to GUGA program. Many alternatives are possible for performing an MRCI calculation as shown in the next example below, in which the reference space to perform the CI is multiconfigurational:
The MRCI program also allows the calculation of electronic energies using the ACPF method. Another MOLCAS program, CPF, offers the possibility to use the CPF, MCPF, and ACPF methods with a single reference function. The required input is quite similar to that for the MRCI program:
Finally, MOLCAS can also perform closed- and open-shell coupled cluster calculations at the CCSD and CCSD(T) levels. These calculations are controlled by the CCSDT program, whose main requirement is that the reference function has to be generated with the RASSCF program. The following input is required to obtain a CCSD(T) energy for the CH4 molecule:
Since this is a closed-shell calculation, the RASSCF input computes a simple RHF wave function with zero active electrons and orbitals using keywords OutOrbitals and Canonical. The MOTRA must include the keyword JobIph to extract the wave function information from file JOBIPH which is automatically generated by RASSCF. Finally, the keywork CCT in program CCSDT leads to the calculation of the CCSD(T) energy using the default algorithms. The CCSDT program in MOLCAS is especially suited to compute open-shell cases. The input required to obtain the electronic energy of the CH4+ cation with the CCSD(T) method is:
where the RASSCF program generated the proper Restricted Open-Shell Hartree-Fock (ROHF) reference. Different levels of spin adaptation are also available. If solvent effects are desired, MOLCAS includes two models: Kirkwood and PCM. Adding a solvent effect to a ground state at HF, DFT, or CASSCF levels, simply requires the inclusion of the keyword RF-input within the input for the SEWARD which calculates a self-consistend reaction field.
Other programs such as CASPT2, RASSI, and MOTRA require that the reaction field is included as a perturbation with keyword RFPErturbation. In the next example the correction is added at both the CASSCF and CASPT2 levels.
Notice that the tesserae of the average area in the PCM model (keyword
has been changed to the value required for acetone by the keyword Aare,
while the default is 0.4 Å2 for water
(see section
Next: 5.2 Optimizing geometries Up: 5. Problem Based Tutorials Previous: 5. Problem Based Tutorials |
 , including the file
SCF.energy.CH4 described below.
, including the file
SCF.energy.CH4 described below.
![[*]](crossref.png)
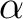 and
and 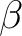 orbital occupations in two ways.
orbital occupations in two ways.

