
























MOLCAS manual: Next: 8.4 casvb Up: 8. Programs Previous: 8.2 averd
|
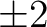 kcal/mol. These results have been obtained with saturated basis sets and all
valence electrons active. The use of smaller basis sets and other types of
active spaces may, of course, affect the error.
kcal/mol. These results have been obtained with saturated basis sets and all
valence electrons active. The use of smaller basis sets and other types of
active spaces may, of course, affect the error.
![[*]](crossref.png)
 , the
number of irreps of the point group.
, the
number of irreps of the point group.
| File | Contents |
| PT2ORB | Molecular orbitals. |
8.3.3 Input
This section describes the input to the CASPT2 program, starting with its name:
&CASPT2
8.3.3.1 Keywords
| Keyword | Meaning |
| TITLe | This keyword is followed by one title line. |
| MULTistate | Enter number of root states, and a list of which CI vector from
the CASSCF calculation to use for each state, for example ``2 1 2''
would specify the first and second root.
Also used for single-state calculations, when the root state is not
the ground state, for example ``1 2'' would specify the second root.
The special value ``all'' can be used if all the states included
in the CASSCF orbital optimization (keyword CIRoot in RASSCF)
are desired.
|
| IPEAshift | This shift corrects the energies of the active orbitals and is
specified in atomic units. It will be weighted by a function of the
diagonal density matrix element 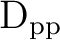 .
This option is used to modify the standard definition of the
zeroth order Hamiltonian (H0), which includes an IPEA shift of 0.25
[28]. The modification of H0 has been introduced (Nov 2005) to
reduce the systematic error which leads to a relative overestimation of the
correlation energy for open shell system. It also reduces the intruder problems.
Default is to use an IPEA shift of 0.25. .
This option is used to modify the standard definition of the
zeroth order Hamiltonian (H0), which includes an IPEA shift of 0.25
[28]. The modification of H0 has been introduced (Nov 2005) to
reduce the systematic error which leads to a relative overestimation of the
correlation energy for open shell system. It also reduces the intruder problems.
Default is to use an IPEA shift of 0.25.
|
| IMAGinary | Add an imaginary shift to the external part of the zero order Hamiltonian. The correlation energy computed is the real part of the resulting complex perturbation energy. Also, a corrected value, obtained by Hylleraas' variational formula, is computed. See Ref. [32]. As with the real shift, this option is used to eliminate intruder problems. |
| SHIFt | Add a shift to the external part of the zero order Hamiltonian. See Refs. [32,30,26]. In addition to the conventionally computed second order energy value, another energy obtained by Hylleraas' variational formula is computed. This energy is then very close to the unshifted energy, except close to singularities due to intruders. This option should only be used to eliminate intruder state problems. |
| AFREeze | This keyword is used to select atoms for defining the correlation orbital
space for the CASPT2 calculation. Assume that you have a large molecule where
the activity takes place in a limited region (the active site). It could be a
metal atom with its surrounding ligands. You can then use this option to reduce
the size of the CASPT2 calculation by freezing and deleting orbitals that have
only a small population in the active site. An example: The cobalt imido complex
CoIII(nacnac)(NPh) has 43 atoms. The active site was cobalt and the
surrounding ligand atoms. Using the AFRE option reduces the time for the CASPT2
calculation from 3 hrs to 3 min with a loss of accuracy in relative energies for
24 electronic states of less than 0.1 eV. The first line after the keyword
contains the number of selected atoms then the selection thresholds (the
recommended value is 0.1 or less). An additional line gives the names of the
atoms as defined in the Seward input. Here is a sample input for the cobalt
complex mentioned above.
AFREeze 6 0.10 0.00 Co N1 N2 C5 C6 C7 This input means that inactive orbitals with less than 0.1 of the density on the active sites will be frozen, while no virtual orbitals will be deleted. |
| LOVCaspt2 | ``Freeze-and-Delete'' type of CASPT2, available only in connection with Cholesky or RI.
Needs (pseudo)canonical orbitals from RASSCF. An example of input for the keyword LOVC is the following:
LovCASPT2 0.3 DoMP2 (or DoEnv) In this case, both occupied and virtual orbitals (localized by the program) are divided in two groups: those mainly located on the region determined (automatically) by the spatial extent of the active orbitals (``active site''), and the remaining ones, which are obviously ``outside'' this region. The value of the threshold (between 0 and 1) is used to perform this selection (in the example, 30% of the gross Mulliken population of a given orbital on the active site). By default, the CASPT2 calculation is performed only for the correlating orbitals associated with the active site. The keyword DoMP2 is optional and forces the program to perform also an MP2 calculation on the ``frozen region''. Alternatively, one can specify the keyword VirAll in order to use all virtual orbitals as correlating space for the occupied orbitals of the active site. A third possibility is to use the keyword DoEnv to compute the energy of the environment as total MP2 energy minus the MP2 energy of the active site. |
| FNOCaspt2 | Performs a Frozen Natural Orbital (FNO) CASPT2 calculation, available only in combination with Cholesky or RI integral representation.
Needs (pseudo)canonical orbitals from RASSCF. An example of input for the keyword FNOC is the following:
FNOCaspt2 0.4 DoMP2 The keyword FNOC has one compulsory argument (real number in ]0,1]) specifying the fraction of virtual orbitals (in each irrep) to be retained in the FNO-CASPT2 calculation. The keyword DoMP2 is optional and used to compute the (estimated) correction for the truncation error. |
| FOCKtype | Use an alternative Fock matrix. The default Fock matrix is described in [21,22] and the other original CASPT2 references. The three different modifications named G1, G2 and G3 are described in [29]. Note: from 6.4 it is not recommended to use this keyword but stay with the IPEA modified H0, which is default. |
| FROZen | This keyword is used to specify the number of frozen orbitals, i.e. the orbitals that are not correlated in the calculation. The next line contain the number of frozen orbitals per symmetry. The default is to freeze the max of those that were frozen in the RASSCF calculation and the deep core orbitals. The frozen orbitals are always the first ones in each symmetry. |
| DELEted | This keyword is used to specify the number of deleted orbitals, i.e. the orbitals that are not used as correlating orbitals in the calculation. The next line contain the number deleted orbitals per symmetry. The default is to delete those that were deleted in the RASSCF calculation. The deleted orbitals are always the last ones in each symmetry. |
| DENSity | Computes the full density matrix from the first order wave function, rather than approximated as is the (faster) default option. Used to compute CASPT2 properties, such as dipole moments, etc. |
| RFPErt | This keyword makes the program add reaction field effects to the energy calculation. This is done by adding the reaction field effects to the one-electron Hamiltonian as a constant perturbation, i.e. the reaction field effect is not treated self consistently. The perturbation is extracted from RUNOLD, if that file is not present if defaults to RUNFILE. |
| RLXRoot | Specifies which root to be relaxed in a geometry optimization of a multi state CASPT2 wave function. Defaults to the highest root or root defined by the same keyword in the RASSCF module. |
| THREsholds | On next line, enter two thresholds: for removal of zero-norm components in the first-order perturbed wave function, and for removal of near linear dependencies in the first-order perturbed wave function. Default values are 1.0d-10 and 1.0d-08 respectively. |
| MAXIter | On next line, enter the maximum allowed number of iterations in a procedure for solving a system of linear equations using a conjugate gradient method. Default is 20. A gradient norm is reported. This gradient is a residual error from the CASPT2 equation solution and should be small, else the number of iterations must be increased. |
| CONVergence | On next line, enter the convergence threshold for the procedure described above. The iterative procedure is repeated until the norm of the residual (RNORM) is less than this convergence threshold. Default is 1.0d-06. |
| NOMIx | Normally, a Multi-State CASPT2 calculation produces new jobiph file named JOBMIX. It has the same CASSCF wave functions as the original ones, except that those CI vectors that was used in the Multi-State CASPT2 calculation have been mixed, using the eigenvectors of the effective Hamiltonian matrix as transformation coefficients. Keyword NOMIX prevents creation of this JOBMIX file. |
| NOMUlt | This keyword removes the multi-state part of the calculation and only runs a series of independent CASPT2 calculations for the roots specified by the MULTistate keyword. Useful when many roots are required, but multi-state is not needed, or desired. Note that a JOBMIX file is produced anyway, but the vectors will not be mixed, and the energies will be single-state CASPT2 energies. |
| ONLY | This keyword requires the MULTistate keyword, and is followed by an integer specifying one of the roots. In a Multistate calculation, it requests to compute the energy of only the specified root. However, the effective Hamiltonian coupling terms between this root and all the others included in the Multistate treatement will be computed and printed out. This output will be used in a subsequent calculation, in conjuction with the EFFE keyword. |
| EFFE | This keyword requires the MULTistate keyword. It is followed by a matrix of real numbers, specifying the effective Hamiltonian couplings, as provided in a previous calculation using the ONLY keyword. In a Multistate calculation over, e.g., 3 states, 3 separate calculations with the ONLY keyword will be performed, possibly on separate computing nodes, so as to speed up the overall process. The three couplings vectors will be given to the EFFE keyword in matrix form, i.e. the first column is made by the couplings of the first computed root, etc. The program will then quickly compute the Multistate energies. |
| NOORbitals | In calculations with very many orbitals, use this keyword to skip the printing of the MO orbitals. |
| PROPerties | Normally, a CASPT2 calculation does not produce any density matrix, natural orbitals or properties in order to save time and memory (especially for large calculations). Keyword PROP activates these calculations, at the expense of (some) extra time and memory (especially if used together with the DENS keyword). |
| NOTRansform | This keyword specifies that the wave function should not be transformed to use quasi-canonical orbitals, even if CASPT2 does not know if this was done or not and by default would do such a transformation. Effectively, the Fock matrix is replaced by a diagonal approximation in the input orbital system. |
| TRANsform | This keyword specifies that the wave function should be transformed to use pseudo-canonical orbitals, even if this was specified as option to the CASSCF calculation and should be unnecessary. (Default is: to transform when necessary, and not else.) |
| OFEMbedding | Adds an Orbital-Free Embedding potential to the hamiltonian. Available only in combination with Cholesky or RI integral representation. No arguments required. The runfile of the environment subsystem (AUXRFIL) must be available. |
| GHOStdelete | Excludes from PT2 treatment orbitals localized on ghost atoms. A threshold for this selection must be specified. |
| OUTPut | Use this keyword, followed by any of the words BRIEF, DEFAULT, or LONG, to control the extent of orbital listing. BRIEF gives a very short orbital listing, DEFAULT a normal output, and LONG a detailed listing. |
| PRWF | This keyword is used to specify the threshold for printing the CI coefficients, default is 0.05. |
| PRSD | This keyword is used to request that not only CSFs are printed with the CI coefficients, but also the determinant expansion. |
| CHEMps2 | Activate DMRG-CASPT2 calculation with MOLCAS–CheMPS2 interface. The keyword 3RDM must be used in RASSCF. The program will skip the calculations of the n-particle reduced density matrix. Note that multistate calculations are not supported, the calculation will run but produce wrong CASPT2 total energy. Always specify MULTi = 1 iroot, where iroot is the root index. |
| CUMUlant | Activate DMRG-cu(4)-CASPT2 calculation with MOLCAS–Block interface. The keyword 3RDM must be used in RASSCF. The program will skip the calculations of the 3-particle reduced density matrix and approximate the 4-particle reduced density matrix. |
The given default values for the keywords Convergence and Thresholds normally give a second order energy which is correct in eight decimal places.
8.3.3.2 Input example
&CASPT2
Title
The water molecule
Density matrix
The CASPT2 energy and density matrix is computed for the water molecule with the O(1s) orbital frozen. The standard IPEA-H0 is used.
Input example for SS-DMRG-CASPT2 with MOLCAS–CheMPS2 interface
&RASSCF
Title = Water molecule. Ground state
Spin = 1
Symmetry = 1
Inactive = 2 0 1 0
Ras2 = 2 2 0 0
DMRG = 500
3RDM
&CASPT2
CHEMps2
Input example for SA-DMRG-CASPT2 with MOLCAS–CheMPS2 interface
&RASSCF
Title = Water molecule. Averaging two states
Spin = 1
Symmetry = 1
Inactive = 2 0 1 0
Ras2 = 2 2 0 0
CIROot = 2 2 1
DMRG = 500
&RASSCF
Title = Ground state
Spin = 1
Symmetry = 1
Inactive = 2 0 1 0
Ras2 = 2 2 0 0
CIROot = 1 1 ; 1
DMRG = 500
LUMOrb
CIONly
3RDM
&CASPT2
CHEMps2
MULTistate = 1 1
&RASSCF
Title = First excited state
Spin = 1
Symmetry = 1
Inactive = 2 0 1 0
Ras2 = 2 2 0 0
CIROot = 1 2 ; 2
DMRG = 500
LUMOrb
CIONly
3RDM
&CASPT2
CHEMps2
MULTistate = 1 2
Next: 8.4 casvb Up: 8. Programs Previous: 8.2 averd

