
























MOLCAS manual: Next: 8.7 cht3 Up: 8. Programs Previous: 8.5 ccsdt
The CHCC is a Closed-Shell Coupled-Clusters Singles and Doubles
program based exclusively on the Cholesky (or RI) decomposed 2-electron integrals
aimed towards calculation of large systems on highly parallel architectures. Use of
point-group symmetry is not implemented. Main advantage compared to the
CCSDT module in MOLCAS is in its more efficient parallelization and
dramatically lowered memory (and eventually disk) requirements.
|
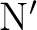 , should be as small as
possible, because increasing of the segmentation brings in more CPU and I/O overhead.
Furthermore, blocking can be ``fine tuned'' by, so called, ``small'' segmentation (SMALl),
, should be as small as
possible, because increasing of the segmentation brings in more CPU and I/O overhead.
Furthermore, blocking can be ``fine tuned'' by, so called, ``small'' segmentation (SMALl), 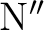 ,
which affects only the (typically) most demanding
,
which affects only the (typically) most demanding 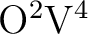 scaling
terms. The ``large'' segmentation can range from 1 to 32, ``small'' segmentation from 1 to 8, but
their product, i.e. ``large x small'' must be no more than 64.
scaling
terms. The ``large'' segmentation can range from 1 to 32, ``small'' segmentation from 1 to 8, but
their product, i.e. ``large x small'' must be no more than 64.
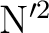 terms scaling as
terms scaling as
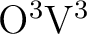 and 1/2
and 1/2 ![[*]](crossref.png)
| File | Contents |
| L0xxxx, L1xxxx, L2xxxx | MO-transformed Cholesky vectors |
| T2xxxx | T2 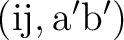 excitation amplitudes excitation amplitudes
|
| RstFil | Communication file containing T1 amplitudes, restart informations, etc. |
8.6.3 Input
The input for each module is preceded by its name like:
&CHCC
Optional keywords
| Keyword | Meaning |
| TITLe | This keyword is followed by one title line. |
| FROZen | Integer on the following line specifies number of inactive occupied orbitals in the CCSD calculation. (Default=0) |
| DELEted | Integer on the following line specifies number of inactive virtual orbitals in the CCSD calculation. (Default=0) |
| LARGe | Integer on the following line specifies the main segmentation of the virtual orbitals. Value must be between 1 (no segmentation) and 32. Product of Large and Small segmentation must be lower than 64. (Default=1) |
| SMALl | Integer on the following line specifies the auxiliary segmentation of the virtual orbitals. Value must be between 1 (no segmentation) and 8. Product of Large and Small segmentation must be lower than 64. Small segmentation doesn't generate extra parallel tasks. (Default=1) |
| CHSEgmentation | Integer on the following line specifies the block size of the auxiliary (Cholesky/RI) index. Value must lower than the minimal dimension of the auxiliary index on each computational node. (Default=100) |
| MHKEy | Integer on the following line specifies if library BLAS (MHKEy=1) or hard-coded fortran vector-vector, matrix-vector and matrix-matrix manipulation is used. (Default=1) |
| NOGEnerate | This keyword specifies that the pre-CCSD steps (regeneration of integrals from the Cholesky/RI vectors, etc.) are skipped. (Default=OFF) |
| ONTHefly | This keyword specifies that all integral types scaling steeper then O2V2 are generated "on-the-fly" from the Cholesky/RI vectors. Use of this keyword leads to dramatically savings of the disk resources, but leads to significant arithmetic overhead. Keywords "ONTHefly" and "PRECalculate" are mutually exclusive. (Default=OFF) |
| PRECalculate | This keyword specifies that all integral are precalculated before the CCSD iterative procedure starts. Use of this keyword leads to significant consumption of the disk space, especially is single-processor runs. (Default=ON) |
| NODIstribute | This keyword (in combination with the "PRECalculate" keyword) specifies that all
integral are stored on each computational node. In case of all integrals being
stored on each node, extra permutation symmetry can be applied, thus leading to
significant savings of the disk space. However, in case of massively parallel runs
(i.e. more than  8 nodes), savings from keeping only subset of integrals
required on particular node are more significant than savings due to permutational
symmetry. (Default=OFF) 8 nodes), savings from keeping only subset of integrals
required on particular node are more significant than savings due to permutational
symmetry. (Default=OFF)
|
| JOINlkey | The parameter on the following line specifies, which algorithm is used for
precalculation and of the integrals in parallel run. In parallel runs, SEWARD
produces AO Cholesky/RI vectors segmented in auxiliary index over
parallel nodes. Depending on the network bandwidth and computational power
of each node, different algorithms can lead to optimal performance.
Following options are available:
0 - None: no cumulation of Cholesky/RI vectors is needed (debug only). 1 - Minimal: Cholesky/RI vectors are cumulated prior to integral precalculation. Low network bandwidth is required. 2 - Medium: O2V2 integrals are generated from local Cholesky/RI vectors and cumulated along with the Cholesky/RI vectors afterwards. Other integrals are calculated from cumulated intermediates. 3 - Full: All integrals are generated from local Cholesky/RI vectors and cumulated afterwards. High network bandwidth is required. (Default=2) |
| MAXIterations | Integer on the following line specifies maximum number of CCSD iteration (Default=40) |
| RESTart | This keyword specifies that CCSD calculation is restarted from previous run. This keyword is currently under development, thus disabled. (Default=OFF) |
| THREshold | Double precision floating point number on the following line specifies the convergence threshold for the CCSD correlation energy. (Default=1.0d-6) |
| PRINtkey | The integer on the following line specifies the print level in output
1 - Minimal 2 - Minimal + timings of each step of the CCSD iterations 10 - Debug (Default=1) |
| END of input | This keyword indicates that there is no more input to be read. |
&CHCC &END
Title
Benzene dimer
Frozen
12
Deleted
0
Large
4
Small
2
CHSEgment
100
Precalculate
Join
2
Maxiter
50
Threshold
1.0d-6
Print
2
End of Input
Next: 8.7 cht3 Up: 8. Programs Previous: 8.5 ccsdt

