
























MOLCAS manual: Next: 8.13 espf (+ QM/MM interface) Up: 8. Programs Previous: 8.11 dynamix
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| File | Contents |
| EMBQ.INP | This file contains keywords and control parameters for the EMBQ program including information about the crystal lattice cell and atoms of the QM cluster. No point group symmetry is assumed. |
8.12.2.2 Intermediate files
All the intermediate files are created, used and removed automatically.
8.12.2.3 Output files
In all output files coordinates are given in Ångstroms and the values of charges – in atomic units.| File | Contents |
| EMBQ_cell.xyz | Lattice cell: coordinates and ionic charges of the lattice cell atoms. No point group symmetry is assumed. Format: XYZ. |
| EMBQ_cell+Q.xyz | Modified cell: coordinates and charges of the lattice cell atoms and complementary point charges generated by EMBQ. Format: XYZ. Note that the lattice atoms and complementary charges may coincide. |
| EMBQ_ncQ.xyz | Coordinates and charges of all species of the nano-cluster constructed using the modified unit cell. Format: XYZ. |
| EMBQ_ncQ.dat | Coordinates and charges of all species of the nano-cluster constructed using the modified unit cell. Format: 4 columns containing Cartesian coordinates and the value of the charge. |
| EMBQ_ncQ-QM.xyz | Coordinates and charges of all species of the nano-cluster without atoms of the QM cluster (if specified). Format: XYZ. |
| EMBQ_ncQ-QM.dat | Coordinates and charges of all species of the nano-cluster without atoms of the QM cluster (if specified). Format: 4 columns containing Cartesian coordinates and the value of the charge. |
| EMBQ_elpot.dat | Coordinates and charges of the nano-cluster, distance from the centre of the nano-cluster to each centre, centre number, on-site electrostatic potential and components of the field. |
8.12.3 Input
Description of the input to EMBQ is below. The keywords
are always significant to four characters, but in order to make the
input more transparent, it is recommended to use the full keywords.
The EMBQ program section of the MOLCAS input is bracketed by
a preceding dummy namelist reference
&EMBQ
End of Input
Argument(s) to a keyword are always supplied on the next line of the input file, except explicitly stated otherwise.
8.12.3.1 Compulsory keywords
| Keyword | Meaning |
| ELMOment | Keyword, followed by a single integer, which specifies the largest electric multipole to be eliminated. A non-negative integer has to be supplied as argument. Default value is 0. |
| UCVEctors | Specifies parameters of the crystal cell (in Å).
Three lines, containing three real numbers each, have to be supplied:
1st line – components of the cell vector 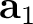 ; ;
2nd line – components of the cell vector 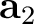 ; ;
3rd line – components of the cell vector 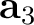 . .
|
| UCV1 | Keyword, followed by three real numbers.
Specifies components of the crystallographic cell vector (in Å).
Can be used as an alternative to keyword UCVEctors.
Should be used together with keywords UCV2 and UCV3.
There is no default value.
|
| UCV2 | Keyword, followed by three real numbers.
Specifies components of the crystallographic cell vector (in Å).
Can be used as an alternative to keyword UCVEctors.
Should be used together with keywords UCV1 and UCV3.
There is no default value.
|
| UCV3 | Keyword, followed by three real numbers.
Specifies components of the crystallographic cell vector (in Å).
Can be used as an alternative to keyword UCVEctors.
Should be used together with keywords UCV1 and UCV2.
There is no default value.
|
| UCAToms | Keyword, followed by a single integer equal to the number of atoms in the lattice cell and a list of the corresponding coordinates (in Å) and ionic charges (in atomic units). |
8.12.3.2 Optional keywords
| Keyword | Meaning | ||||||||||||||||||||||||||||||||||||||||
| TETRahedra | Keyword, followed by a single line containing eight integers equal to either 0 or 1.
Specifies which tetrahedra are used to complement the crystal cell with EMBQ charges.
Each integer correspond to a single tetrahedron associated with a single corner of the crystal cell.
The tetrahedron is used if the corresponding parameter equals to 1 and not used if it equals to 0.
Default: use all eight tetrahedra.
Orientation of the tetrahedra are determined by their axes, which are either parallel (+) or anti-parallel (–) to the cell vectors
| ||||||||||||||||||||||||||||||||||||||||
| SHIFt | Keyword, followed by a single real number. Specifies the shift (in fractional coordinates) of the tetrahedra from the cell corners outwards. Default value is zero. | ||||||||||||||||||||||||||||||||||||||||
| NANOcluster | Keyword followed by two input lines.
The first line contains a single integer number (n) which specifies the shape of a nano-cluster generated using the modified cells. Possible values of n:
1 – to generate a cubic nano-cluster 2 – to generate a block nano-cluster 3 – to generate a spherical nano-cluster. The size of the nano-cluster is defined in the following line. The number of input parameters depends on the shape of the nano-cluster. For a cube, provide one integer k to generate a nano-cluster of (2k+1)3 unit cells. For a block, provide six integers k1, k2, m1, m2, n1, n2 to generate a nano-cluster of
(k2 - k1 + 1) x ( m2 - m1 + 1) x (n2 - n1 + 1)
unit cells. For a sphere, provide one real number to generate a nano-cluster of radius R (in Å). This keyword can be used instead of keywords NCCube, NCBLock, and NCSPhere. Note that only one nano-cluster will be generated. Default: the nano-cluster is not generated. | ||||||||||||||||||||||||||||||||||||||||
| NCCUbe | Keyword, followed by a single integer number k.
Specifies the shape and size of the nanocluster constructed from the modified unit cells.
The nanocluster is generated as a block of
(2k+1) x (2k+1) x (2k+1)
cells along the lattice vectors , , and , respectively.
Default: the nanocluster is not generated.
| ||||||||||||||||||||||||||||||||||||||||
| NCBLock | Keyword followed by six integers: k1 k2 m1 m2 n1 n2.
Specifies the shape and size of the nanocluster constructed from the modified unit cells.
The nanocluster is generated as a block of
(k2-k1+1) x (m2-m1+1) x (n2-n1+1)
cells along the lattice vectors , , and , respectively.
Default: the nanocluster is not generated.
| ||||||||||||||||||||||||||||||||||||||||
| NCSPhere | Keyword followed by a single real number. Specifies the shape and radius (in Å) of the nano-cluster constructed from the modified unit cells. Default: the nanocluster is not generated. | ||||||||||||||||||||||||||||||||||||||||
| Keyword, followed by a single integer number.
Specifies the general print level:
0 – minimal print out; 1 – intermediate print out; 2 – full print out. Default: use the global MOLCASprint level. | |||||||||||||||||||||||||||||||||||||||||
| CALCulate | Keyword, followed by a single integer number.
Requests calculation of the electrostatic potential and field at all centres of the nano-cluster.
Possible values are:
0 – calculate neither the potential nor components of the field vector (default); 1 – calculate the potential only; 2 – calculate the potential and components of the field vector. | ||||||||||||||||||||||||||||||||||||||||
| QMCLuster | Keyword, followed by a single integer equal to the number of atoms in the QM cluster and a list of the corresponding Cartesian coordinates (in Å). Specifies geometrical structure of the QM cluster. Default: number of the QM cluster atoms is zero. | ||||||||||||||||||||||||||||||||||||||||
8.12.3.3 Limitations
The largest electric moment ELMOment is limited to 10.Number of atoms in UCAToms is limited to 1000.
Number of atoms in QMCLuster is limited to 1000.
Tetrahedra in TETRahedra are oriented so as three of their edges are parallel to the cell vectors.
The value of SHIFt is the same for all tetrahedra.
8.12.3.4 Input example
&EMBQ &END
Elmoment
4 Largest moment to eliminate
Tetrahedra
1 1 1 1 0 0 0 0 Use the tetrahedra (if 1) or not (if 0)
Shift
0.5 Shift the tetrahedra from the corner sites outward by this value
Nanocluster
3 Shape of the nano-cluster (1 -- cube, 2 -- cuboid, 3 -- sphere)
30.0 Size of the nano-cluster. Here, radius of the sphere (in Å).
2 Printing level
Calculate
2 Calculate electrostatic potential and its derivatives.
UCvectors
4.593730 0.000000 0.000000 Unit cell vector a1 (in Å)
0.000000 4.593730 0.000000 Unit cell vector a2 (in Å)
0.000000 0.000000 2.958120 Unit cell vector a3 (in Å)
UCatoms
6 Number of atoms in the cell
0.000000000 0.000000000 0.000000000 4.0
2.296865000 2.296865000 1.479060000 4.0
1.402465769 1.402465769 0.000000000 -2.0
3.699330769 0.894399231 1.479060000 -2.0
3.191264231 3.191264231 0.000000000 -2.0
0.894399231 3.699330769 1.479060000 -2.0
QMatoms
4 Number of atoms in the QM cluster
1.402465769 1.402465769 0.000000000
2.296865000 2.296865000 -1.479060000
2.296865000 2.296865000 1.479060000
0.000000000 0.000000000 0.000000000
End of Input
Next: 8.13 espf (+ QM/MM interface) Up: 8. Programs Previous: 8.11 dynamix

