
























MOLCAS manual: Next: 8.26 LoProp Up: 8. Programs Previous: 8.24 GUGADRT
|
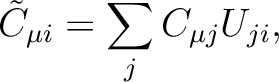 |
(8.9) |
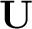 is unitary (i.e. orthogonal for real orbitals).
The same is true for the inactive or active orbitals in a CASSCF wave function.
Whereas the Pipek-Mezey[61],
Boys[62,63], and
Edmiston-Ruedenberg[64] procedures define
through an iterative maximization of a localisation functional,
the Cholesky orbitals are simply defined through the Cholesky decomposition
of the one-electron density, i.e.
is unitary (i.e. orthogonal for real orbitals).
The same is true for the inactive or active orbitals in a CASSCF wave function.
Whereas the Pipek-Mezey[61],
Boys[62,63], and
Edmiston-Ruedenberg[64] procedures define
through an iterative maximization of a localisation functional,
the Cholesky orbitals are simply defined through the Cholesky decomposition
of the one-electron density, i.e.
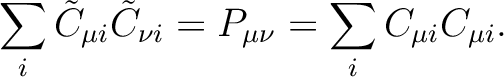 |
(8.10) |
Although these localisation schemes are mostly meant for localising occupied orbitals (except for PAOs which are defined for the virtual orbitals), the LOCALISATION program will attempt to localise any set of orbitals that the user specifies. This means that it is possible to mix occupied and virtual orbitals and thereby break the Hartree-Fock invariance. The default settings, however, do not break the invariance.
For Pipek-Mezey, Boys, and Edmiston-Ruedenberg localisations, iterative
optimizations are carried out. We use
the 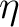 -steps of Subotnik et al.[66] for
Edmiston-Ruedenberg, whereas the traditional Jacobi sweeps (consecutive
two-by-two orbital rotations)[61,66]
are employed for the Pipek-Mezey and Boys schemes.
-steps of Subotnik et al.[66] for
Edmiston-Ruedenberg, whereas the traditional Jacobi sweeps (consecutive
two-by-two orbital rotations)[61,66]
are employed for the Pipek-Mezey and Boys schemes.
8.25.2 Dependencies
The LOCALISATION program requires the one-electron integral file
ONEINT and the communications file RUNFILE,
which contains, among other data, the
basis set specifications processed by GATEWAY and SEWARD.
In addition, the Edmiston-Ruedenberg procedure requires the presence
of Cholesky decomposed two-electron integrals produced by SEWARD.
8.25.3 Files
Below is a list of the files that are used/created by the program LOCALISATION.
8.25.3.1 Input files
LOCALISATION will use the following input files: ONEINT, RUNFILE, INPORB. For Edmiston-Ruedenberg localisation, it also needs CHVEC, CHRED and CHORST files (for more information see![[*]](crossref.png) ).
).
8.25.3.2 Output files
| File | Contents |
| LOCORB | Localised orthonormal orbital output file. Note that LOCORB contains all orbitals (localised as well as non-localised according to the input specification). |
| DPAORB | Linearly dependent nonorthonormal projected atomic orbital output file (only produced for PAO runs). |
| IPAORB | Linearly independent nonorthonormal projected atomic orbital output file (only produced for PAO runs). |
| RUNFILE | Communication file for subsequent programs. |
| MD_LOC | Molden input file for molecular orbital analysis. |
8.25.4 Input
Below follows a description of the input to LOCALISATION.
The LOCALISATION program section of the MOLCAS input is bracketed by
a preceding program reference
&LOCALISATION
8.25.4.1 Optional general keywords
| Keyword | Meaning |
| FILEorb | The next line specifies the filename containing the input orbitals that will be localised. By default a file named INPORB will be used. |
| NORBitals | The following line specifies the number of orbitals to localise in each irreducible representation. The default is to localise all occupied orbitals as specified in the INPORB input file, except for PAO runs where all the virtual orbitals are treated by default. |
| NFROzen | The following line specifies the number of orbitals to freeze in each irreducible representation. The default is not to freeze any orbitals, except for the localisations of the virtual space (see keywords PAO and VIRTual) where the default is to freeze all occupied orbitals (occupation number different from zero, as reported in the INPORB file). |
| FREEze | Implicit frozen core option. The default is not to freeze any orbitals, except for the localisations of the virtual space (see keywords PAO and VIRTual) where the default is to freeze all occupied orbitals (occupation number different from zero, as reported in the INPORB file). The definition of core orbitals is taken from program SEWARD. |
| OCCUpied | Requests that the occupied orbitals should be localised. This is the default except for PAO where the default is virtual. |
| VIRTual | Requests that the virtual orbitals should be localised. The default is to localise the occupied orbitals, except for PAO where the default is virtual. |
| ALL | Requests that all orbitals should be localised. The default is to localise the occupied orbitals, except for PAO where the default is virtual. |
| PIPEk-Mezey | Requests Pipek-Mezey localisation. This is the default. |
| BOYS | Requests Boys localisation. The default is Pipek-Mezey. |
| EDMIston-Ruedenberg | Requests Edmiston-Ruedenberg localisation. The default is Pipek-Mezey. Note that this option requires that the Cholesky (or RI/DF) representation of the two-electron integrals has been produced by SEWARD. |
| CHOLesky | Requests Cholesky localisation (non-iterative). The default is Pipek-Mezey. This and PAO are the only options that can handle point group symmetry. The decomposition threshold is by default 1.0d-8 but may be changed through the THREshold keyword. |
| PAO | Requests PAO localisation (non-iterative) using Cholesky decomposition to remove linear dependence. The default is Pipek-Mezey. This and Cholesky are the only options that can handle point group symmetry. The decomposition threshold is by default 1.0d-8 but may be changed through the THREshold keyword. |
| SKIP | Leaves the input orbitals unchanged. It is turned off by default. |
| ITERations | The following line specifies the maximum number of iterations to be used by the iterative localisation procedures. The default is 100. |
| THREshold | The following line specifies the convergence threshold used for changes in the localisation functional. The default is 1.0d-6. For Cholesky and PAO methods, it is the decomposition threshold and the default is 1.0d-8. |
| THRGradient | The following line specifies the convergence threshold used for the gradient of the localisation functional. The default is 1.0d-2. |
| THRRotations | The following line specifies the screening threshold used in the Jacobi sweep optimization algorithm. The default is 1.0d-10. |
| CHOStart | Requests that iterative localisation procedures use Cholesky orbitals as initial orbitals. The default is to use the orbitals from INPORB directly. |
| ORDEr | Requests that the localised orbitals are ordered in the same way as the Cholesky orbitals would be. This is mainly useful when comparing orbitals from different localisation schemes. The ordering is done according to maximum overlap with the Cholesky orbitals. The default is not to order. |
| DOMAin | Requests orbital domains and pair domains are set up and analyzed. The default is not to set up domains. |
| THRDomain | The following line specifies two thresholds to be used in defining orbital domains. The first is the Mulliken population threshold such that atoms are included in the domain until the population (divided by 2) is larger than this number (default: 9.0d-1). The second threshold is used for the Pulay completeness check of the domain (default: 2.0d-2). |
| THRPairdomain | The following line specifies three thresholds to be used for
classifying pair domains: R1, R2, and R3. (Defaults: 1.0d-10,
1.0d1, and 1.5d1.)
If R is the smallest distance
between two atoms in the pair domain (union of the individual orbital
domains), then pair domains are classified according to:
R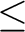 R1: strong pair,
R1<RR2: weak pair,
R2<RR3: distant pair, and
R3<R: very distant pair. R1: strong pair,
R1<RR2: weak pair,
R2<RR3: distant pair, and
R3<R: very distant pair.
|
| LOCNatural orbitals | This keyword is used to select atoms for defining the localised natural
orbitals (LNOs), thus a set of localised orbitals with well-defined occupation numbers.
All other options specified in the LOCALISATION program input apply (e.g., input orbitals,
localisation method, etc.).
On the next line give the number of atoms that identify the region of interest
and the threshold used to select the localised orbitals belonging to this region
(recommended values > 0.2 and < 1).
An additional line gives the names of the (symmetry unique) atoms as defined in the SEWARD input.
The keyword LOCN is used to define suitable occupation numbers for RASSCF active orbitals
that have been localised. It has proven useful in Effective Bond Order (EBO) analysis.
Here is a sample input for a complex containing an iron-iron multiple bond.
LOCN 2 0.3 Fe1 Fe2 In this example, the (localised) orbitals constructed by the LOCALISATION program are subdivided in two groups: those having less than 0.3 total Mulliken population on the two iron atoms, and the remaining orbitals, obviously localised on the iron-iron region. The resulting density matrices for the two subsets of orbitals are then diagonalized separately and the corresponding (localised) natural orbitals written to LOCORB with the proper occupation numbers. Note that the two sets of LNOs are mutually non-orthogonal. |
| LOCCanonical orbitals | This keyword is used to select atoms for defining the localised canonical orbitals (LCOs), thus a set of localised orbitals with well-defined orbital energies (eigenvalues of a local Fock matrix). Please, refer to the analogous keyword LOCN in this manual for more details and input examples. |
8.25.4.2 Limitations
The limitations on the number of basis functions are the same as specified for SEWARD.
8.25.4.3 Input examples
This input is an example of the Boys localisation of the CO molecule. Note that no
symmetry should not be used in any calculation of localised orbitals except for
Cholesky and PAO orbitals.
&GATEWAY
Coord = $MOLCAS/Coord/CO.xyz
Basis = STO-3G
Group = C1
&SEWARD ; &SCF
&LOCALISATION
Boys
This input is an example of the Projected Atomic Orbital localisation of the virtual orbitals of the CO molecule. The threshold for the Cholesky decomposition that removes linear dependence is set to 1.0d-14.
&GATEWAY
Coord = $MOLCAS/Coord/CO.xyz
Basis = STO-3G
Group = C1
&SEWARD ; &SCF
&LOCALISATION
PAO
Threshold = 1.0d-14
This input is an example of the Cholesky localisation (using default 1.0d-8 as threshold for the decomposition) of the valence occupied orbitals of the CO molecule. Orbital domains are set up and analyzed.
&GATEWAY
Coord = $MOLCAS/Coord/CO.xyz
Basis = STO-3G
Group = C1
&SEWARD ; &SCF
&LOCALISATION
Cholesky
Freeze
Domain
Next: 8.26 LoProp Up: 8. Programs Previous: 8.24 GUGADRT

