
























MOLCAS manual: Next: 8.28 mckinley (a.k.a. denali) Up: 8. Programs Previous: 8.26 LoProp
|
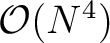 , where N is the number of basis functions)
to the exchange operator matrices
, where N is the number of basis functions)
to the exchange operator matrices 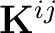 in MO basis
(
in MO basis
(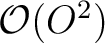 matrices of size V2, where O and V denote the number
of occupied and virtual orbitals, respectively), is either done
conventionally, using the two-electron integral file ORDINT, which
was generated in a previous step by the SEWARD integral code.
matrices of size V2, where O and V denote the number
of occupied and virtual orbitals, respectively), is either done
conventionally, using the two-electron integral file ORDINT, which
was generated in a previous step by the SEWARD integral code.
![[*]](crossref.png)
| File | Contents |
| RUNFILE | File for communication of auxiliary information. |
8.27.4 Input
Below follows a description of the input to MBPT2.
The input for each module is preceded by its name like:
&MBPT2
No compulsory keywords are required for MBPT2. The reference statement mentioned above is sufficient for a default MBPT2 run.
8.27.4.1 Optional keywords
| Keyword | Meaning |
| TITLe | The line following this line is regarded as a title line |
| Specifies the general print level of the calculation. An integer has to be supplied as argument. The default value, 0, is recommended for production calculations. | |
| FREEze | Specifies the total number of frozen occupied orbitals. The lowest-energy occupied orbitals are then automatically identified and frozen. The keyword takes as argument one integer. Incompatible with the FROZen keyword. |
| FROZen | Specifies the number of frozen occupied orbitals in each of the irreducible representations (irreps) of the subgroup of D2h in which the system is represented. The counting of the orbitals follows the increasing orbital energy within each irrep, with those orbitals being frozen first that correspond to lowest orbital energies. The keyword takes as argument nIrrep (# of irreps) integers. Incompatible with the FREEze keyword. Default is to freeze non-valence orbitals. |
| DELEted | Specifies the number of deleted orbitals in each of the irreducible
representations (irreps) of the subgroup of D2h in which the system
is represented. The counting of the orbitals follows the decreasing
orbital energy within each irrep, with those orbitals being deleted first
that correspond to highest orbital energies.
The keyword takes as argument nIrrep (# of irreps) integers.
NOTE: Those orbitals, which have been deleted already in the SCF calculation (cf. SPDElete, OVLDelete of the SCF program description) are never seen by the MBPT2 program and hence are not to be deleted again with the present option. |
| SFROzen | Allows to specify specific orbitals to freeze in each of the irreducible
representations (irreps) of the subgroup of D2h in which the system
is represented. In the 1st line after the keyword the number of orbitals
to freeze for each irrep is specified (nIrrep (# of irreps) integers).
The next  nIrrep lines reference the orbitals to freeze for the
related irrep, following an enumeration of the individual orbitals
of 1, 2, 3, nIrrep lines reference the orbitals to freeze for the
related irrep, following an enumeration of the individual orbitals
of 1, 2, 3, according to
increasing orbital energy. Note that the orbital reference numbers
obey the original ordering and also include those orbitals which
may have been frozen already by the
FROZen or FREEze options. If the corresponding irrep does not contain any
specific orbitals to freeze (i.e. a zero was supplied for this irrep in the
1st line), no line orbital reference input line is supplied for that irrep. according to
increasing orbital energy. Note that the orbital reference numbers
obey the original ordering and also include those orbitals which
may have been frozen already by the
FROZen or FREEze options. If the corresponding irrep does not contain any
specific orbitals to freeze (i.e. a zero was supplied for this irrep in the
1st line), no line orbital reference input line is supplied for that irrep.
|
| SDELeted | Allows to specify specific orbitals to delete in each of the irreducible
representations (irreps) of the subgroup of D2h in which the system
is represented. In the 1st line after the keyword the number of orbitals
to delete for each irrep is specified (nIrrep (# of irreps) integers).
The next nIrrep lines reference the orbitals to delete for the
related irrep, following an enumeration of the individual orbitals
of 1, 2, 3, according to
increasing orbital energy. Note that the orbital reference numbers
obey the original ordering.
If the corresponding irrep does not contain any
specific orbitals to freeze (i.e. a zero was supplied for this irrep in the
1st line), no line orbital reference input line is supplied for that irrep.
|
| GHOStdelete | Excludes from PT2 treatment orbitals localized on ghost atoms. A threshold for this selection must be specified. |
| LUMOrb | Molecular orbital coefficients and energies read from INPORB file rather than RunFile. |
| EREF | Specifies the value of the reference energy. Available only in combination with LumOrb. Default value of the reference energy is set to zero. |
| TEST | If this keyword is specified the input is checked without performing any calculation. |
| T1AM | Singles amplitudes/energy introduced according to Thouless formula. An INPORB file containing MOs different from HF orbitals is required. |
| LOVMp2 | ``Freeze-and-Delete'' type of MP2, available only in connection with Cholesky or RI.
An example of input for the keyword LOVM is the following:
LovMP2 2 0.2 (nCenters,thrs) C1 N (Center labels) DoMP2 In this case, both occupied and virtual orbitals (localized by the program) are divided in two groups: those (A) mainly located on the two (symmetry independent) centers C1 and C2, and the remaining ones (B), which are obviously ``outside'' this region. The value of the threshold (between 0 and 1) is used to perform this selection (in the example, 20% of the gross Mulliken population of a given orbital on the specified atoms). By default, the MP2 calculation is performed only for the correlating orbitals associated with the region A (``active site''). The keyword DoMP2 is optional and forces the program to perform also an independent MP2 calculation on the ``frozen region'' (B). Alternatively, one can specify the keyword VirAll in order to use all virtual orbitals as correlating space for the occupied orbitals of the active site. |
| FNOMp2 | Performs a Frozen Natural Orbital (FNO) MP2 calculation, available only in combination with Cholesky or RI integral representation.
An example of input for the keyword FNOM is the following:
FNOMp2 0.4 DoMP2 The keyword FNOM has one compulsory argument (real number in ]0,1]) specifying the fraction of virtual orbitals (in each irrep) to be retained in the FNO-MP2 calculation. The keyword DoMP2 is optional and used to compute the (estimated) correction for the truncation error. |
| PRPT | Multipole moments (dipoles and quadrupoles) are calculated and printed. The moments are calculated by using a variational one-particle MP2 density matrix. The calculation of the density matrix substantially increases the computational effort compared to an ordinary energy calculation. If the call to MBPT2 is followed by a LoProp call the variational MP2 density matrix will automatically be passed on to that module when this keyword is active. |
| GRDT | Variational one and two-particle MP2 densities are calculated to prepare for analytical gradient calculations. The default for subsequent gradient calculations are changed from numerical to analytical when this keyword is invoked. When using mbpt2 in a slapaf-loop with only C1 symmetry analytical gradients are automatically default and this keyword is not needed. grdt prints Multipole moments and prepare for LoProp in the exact same way as prpt. Use of this keyword therefore makes it redundant (but harmless) to also specify the keyword prpt. |
| NOGRdt | Disables the calculation of variational densities for analytical gradients.
This is useful to cancel the implicit grdt added when using mbpt2
inside a slapaf-loop, if no analytic gradients are actually needed.
Note that using the Numerical keyword in gateway already disables
grdt, so nogrdt is only needed in some advanced situations.
|
8.27.4.2 Optional keywords specific to Cholesky calculations
Observe that these keywords are disregarded if the integrals were not Cholesky decomposed by SEWARD. Furthermore, they are disregarded for algorithm 0 (see below).| Keyword | Meaning |
| CHOAlgorithm | Takes as argument one positive integer specifying the algorithm to use for Cholesky MP2. Options: 0 [generate MO integrals on disk from Cholesky vectors], 1 [compute integrals on-the-fly, minimal operation count, level 2 BLAS], 2 [compute integrals on-the-fly, not minimal operation count, level 3 BLAS], Default is 2. |
| VERBose | Increases printing from the Cholesky MP2 routines, although not by much. Default is (almost) no printing. |
| DECOmpose | Requests Cholesky decomposition of the (ai|bj) integrals. Unless user-defined (see below), the threshold used is identical to that used by SEWARD for decomposing the two-electron integrals. Default is to not decompose. |
| THRCholesky | Specifies the threshold for (ai|bj) Cholesky decomposition. Takes as argument one real number. Default is the threshold used by SEWARD for decomposing the two-electron integrals. |
| NODEcompose | Turns off Cholesky decomposition of the (ai|bj) integrals. Default is to not decompose. |
| SPAN | Specifies the span factor used for (ai|bj) Cholesky decomposition. Takes as argument one real number. Default is the span factor used by SEWARD for decomposing the two-electron integrals. |
| MXQUal | Specifies the max. number of qualified diagonals treated during (ai|bj) Cholesky decomposition. Takes as argument one integer. Default is 10% of the max. rank of (ai|bj), although never more than 200. |
| PRESort | Presort the MO Cholesky vectors according to the batches over occupied orbitals. This will reduce the amount of I/O performed during on-the-fly assembly of the (ai|bj) integrals. This keyword is obsolete. |
8.27.4.3 Limitations
The maximum number of selectively frozen SFRO or selectively deleted orbitals SDEL in each symmetry is limited to 50.The limitations on the number of basis functions are the same as specified for SEWARD.
8.27.4.4 Input example
&MBPT2
Title
H2O: O(9.5/4.2), H(4/2)
* The lowest energy occupied orbital in the repr. no.1 will be frozen in
* MBPT2 calculations. The number of representations is 4 and all zeros
* must be explicitly given
Frozen
1 0 0 0
* Two highest energy external orbitals in the repr. no.3 will be deleted
* in MBPT2 calculations. The number of representations is 4 and all
* zeros must be explicitly given
Deleted
0 0 2 0
* One occupied orbital in symmetry no.1 will be additionally frozen by
* using the SFRO option. Let it be the third SCF occupied orbital in
* this symmetry
sFrozen
1 0 0 0 // Gives the number of frozen orbitals in each symmetry
3 // Gives the frozen orbital reference number in symmetry no. 1
* Two external orbitals in symmetry no.1 and one external orbital in
* symmetry 3 will be deleted. In symmetry 1 let it be the second and
* third external orbitals, and in symmetry 3 the third (already deleted
* in by using the option DELE) external orbital
sDeleted
2 0 1 0 // Gives the number of orbitals to be deleted in each symmetry
2 3 // Gives the reference numbers of external orbitals in sym. 1
3 // Gives the reference number of the external orb. in sym. 3
Next: 8.28 mckinley (a.k.a. denali) Up: 8. Programs Previous: 8.26 LoProp

