
























MOLCAS manual: Next: 8.12 embq Up: 8. Programs Previous: 8.10 dimerpert
|
| File | Contents |
| velocity.xyz | Contains the initial velocities of the MD simulation. |
8.11.2.2 Output files
| File | Contents |
| RUNFILE | Trajectory information such as current time, velocities, etc. are stored in this file. |
| md.xyz | The coordinates for each step of the MD trajectory are saved here. |
| md.energies | The potential, kinetic and total energies are written to this file. In case of multiple electronic states, the energies of all roots are saved. |
8.11.3 Input
This section describes the input syntax of DYNAMIX in the MOLCAS program
package. In general a MD simulation requires a FOREACH loop which contains
several programs to compute the energy and ALASKA for subsequent gradient
computation. The input of the DYNAMIX begins with the program name,
and is followed by the only compulsory keyword VELV which specifies the
velocity Verlet algorithm:
&DYNAMIX
VELV
8.11.3.1 General keywords
| Keyword | Meaning |
| VELVerlet | This keyword specifies the velocity Verlet algorithm [48] to solve Newton's equations of motion. It's the only compulsory keyword in the program. |
| DTime | Defines the 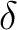 t which is the time step in the MD simulation and which is
used for the integration of Newton's equations of motion.
The program expects the time to be given in floating point
format and in atomic unit of time (1 a.u. of time = 2.42 t which is the time step in the MD simulation and which is
used for the integration of Newton's equations of motion.
The program expects the time to be given in floating point
format and in atomic unit of time (1 a.u. of time = 2.42 10-17 s). (Default = 10). 10-17 s). (Default = 10).
|
| VELOcities | Specifies how the initial velocities are generated.
This keyword is followed by an integer on the next line. The internal
unit of the velocities is [Bohr(a.u. of time)-1].
|
| THERmostat | Regulates the control of the temperature by scaling the velocities. The option
is an integer given on the next line.
|
| TEMPerature | Defines the numerical value of the temperature, which is used together with the Nosé-Hoover chain of thermostats to perform molecular dynamics at constant temperature. (Default = 298.15K) |
| HOP | Enables the trajectory surface hopping algorithm if the integer given in the next line is bigger than 0. The integer also specifies how many non-adiabatic transitions are allowed between electronic states. |
| RESTART | This keyword allows to restart the trajectory at a given time. The time is given on the next line in atomic units. |
| H5RESTART | This keyword allows to restart a trajectory calculation from an HDF5 file. The name of the restart file is given on the next line. |
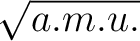
8.11.3.2 Input examples
The following example shows the input for an excited state CASSCF molecular dynamics simulation of a methaniminium cation using the DYNAMIX program. The FOREACH loop allows 1000 steps with 10 a.u. of time step size which leads to a total duration of 242 fs. In the RASSCF program the second root is selected for gradient calculation using the keyword MDRLXR. This input assumes that the a JOBIPH file with orbitals is already given. In each iteration the JOBIPH is updated to achieve a fast convergence of the CASSCF wavefunction. A Nosé-Hoover chain of thermostats, enabled with THERmo= 2, is used to reproduce dynamics at constant temperature, where the initial velocities are taken from a Maxwell-Boltzmann distribution at 300 K.
&GATEWAY
COORD
6
Angstrom
C 0.00031448 0.00000000 0.04334060
N 0.00062994 0.00000000 1.32317716
H 0.92882820 0.00000000 -0.49115611
H -0.92846597 0.00000000 -0.49069213
H -0.85725321 0.00000000 1.86103989
H 0.85877656 0.00000000 1.86062860
BASIS= 3-21G
GROUP= nosym
>> FOREACH ITER in (1 .. 1000)
&SEWARD
>> IF ( $ITER = 1 )
&RASSCF
LUMORB
FileOrb= $Project.GssOrb
Symmetry= 1
Spin= 1
nActEl= 2 0 0
Inactive= 7
RAS2= 2
CIroot= 3 3 1
>> COPY $Project.JobIph $Project.JobOld
>> ENDIF
&RASSCF
JOBIPH; CIRESTART
Symmetry= 1
Spin= 1
nActEl= 2 0 0
Inactive= 7
RAS2= 2
CIroot= 3 3 1
MDRLXR= 2
>> COPY $Project.JobIph $Project.JobOld
&ALASKA
&DYNAMIX
VELVer
DT= 10.0
VELO= 3
THER= 2
TEMP=300
HOP= 1
>> END DO
8.11.4 Dynamixtools
This tool can be found into the Tools/ folder and it will provide some general tools to manage molecular dynamics calculation. At the moment it can be used to generate intial condition (geometries and momenta) following a Boltzmann distribution, based on a frequency calculation. It is working with a freq.molden file (.h5 support coming soon...).
From the command prompt:
>> python3 dynamixtools.py -h
usage: dynamixtools.py [-h] [-s SEED] [-l LABEL] -i I [-b BOL] -t TEMP
optional arguments:
-h, --help show this help message and exit
-s SEED, --seed SEED indicate the SEED to use for the generation of randoms
-l LABEL, --label LABEL
label for your project (default is "geom")
-i I, --input I path of the frequency h5 or molden file
-b BOL, --boltzmann BOL
number of initial condition following boltzmann
distribution (default 1)
-t TEMP, --temperature TEMP
temperature in Kelvin for the initial conditions
Having a water.freq.molden file, this is the command to generate 200 initial condition using 3435432 as seed and a temperature of 300 Kelvin.
>> python3 dynamixtools.py -i water.freq.molden -t 300 -b 200 -s 3435432
Next: 8.12 embq Up: 8. Programs Previous: 8.10 dimerpert

