
























MOLCAS manual: Next: 8.17 gateway Up: 8. Programs Previous: 8.15 falcon
|
| Keyword | Meaning |
| TITLe | Followed by a title line |
| DIPO | Add the dipole moment perturbation operator. By default, the dipole moment integrals are always computed with respect to the center of nuclear charge. The keyword is followed by up to three additional input lines. Each line consists of two entries, the component of the dipole operator and the perturbation length. The component is specified by a single letter (X, Y or Z). |
| QUAD | Add the quadrupole moment perturbation operator. The keyword is followed by at least one additional input line and may be complemented by as many additional lines as needed. Each line consists of two entries, the component of the operator and the perturbation strength. The component is specified by a pair of letters (XX, XY, XZ, YY, YZ or ZZ). By default, the quadrupole moment integrals are calculated with respect to the center of mass. For any other selection the origin of the perturbation operator also needs to be specified by entering a line starting with the string ORIG followed by the coordinates. |
| OCTU | Add the octupole moment perturbation operator. The keyword is followed by at least one additional input line and may be complemented by as many additional lines as needed. Each line consists of two entries, the component of the operator and the perturbation strength. The component is specified by a triple of letters (XXX, XXY, XXZ, XYY, XYZ, XZZ, YYY, YYZ, YZZ, or ZZZ). By default, the octupole moment integrals are calculated with respect to the center of mass. For any other selection the origin of the perturbation operator also needs to be specified by entering a line starting with the string ORIG followed by the coordinates. |
| EFLD | Add the electric field perturbation operator. The keyword is followed by at least two additional input lines and may be complemented by as many additional lines as needed. Each line consists of two entries, the component of the operator and the perturbation strength. The component is specified by a single letter (X, Y or Z). In addition, the origin of the perturbation operator also needs to be specified by entering a line starting with the string ORIG followed by the coordinates. |
| EFGR | Add the electric field gradient perturbation operator. The keyword is followed by at least one additional input line and may be complemented by as many additional lines as needed. Each line consists of two entries, the component of the operator and the perturbation strength. The component is specified by a pair of letters (XX, XY, XZ, YY, YZ or ZZ). In addition, the origin of the perturbation operator also needs to be specified by entering a line starting with the string ORIG followed by the coordinates. |
| RELA | Add the relativistic correction (mass-velocity and one-electron Darwin contact term). The command is followed by one additional line of input specifying the perturbation strength. |
| GLBL | This command marks the beginning of a more general perturbation
description which is not included as a subcommand of the
FFPT command.
This card is followed by as many additional input lines as needed and
is terminated if the next input line starts with a command. Each input
line contains only one perturbation description and three data fields
which are: Label, component and perturbation strength. The label
consists of a character string of length 8 and names the one-
electron integrals produced by SEWARD. The component of
an operator is given as an integer. The last parameter denotes
the strength of a perturbation operator and is given as a real number.
For a list of the available one-electron integral labels refer to
section ![[*]](crossref.png) . .
For example to add Pauli repulsion integrals for
reaction field calculations the input would look like:
|
| SELEctive | With the same localization scheme as used in LOPROP, the perturbation
from FFPT is localized in an orthogonal basis. Then the user can
specify on which basis functions the perturbation should act.
For example, the input
&FFPT DIPO X 0.005 SELECTIVE 2 .true. 1 26 .false. 67 82 .true. 0.5 leads to that the perturbation only acts on densities with (1) both basis function indexes in the set 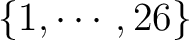 or (2) one index
in the set
while the other is in the set or (2) one index
in the set
while the other is in the set
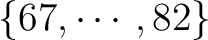 , and in this case the perturbation should be multiplied
by 0.5.; all other densities are unaffected by the perturbation.
We call the former type of subset an atom domain and the latter a bond
domain. Generally, the input structure is this: First line specifies how
many subsets, N, that will be defined. Then follow N lines starting
with a logical flag telling if the subset is an atom domain with the starting
and ending basis function indexes thereafter. N-1 lines follow where the
bond domain is defined in the following way: , and in this case the perturbation should be multiplied
by 0.5.; all other densities are unaffected by the perturbation.
We call the former type of subset an atom domain and the latter a bond
domain. Generally, the input structure is this: First line specifies how
many subsets, N, that will be defined. Then follow N lines starting
with a logical flag telling if the subset is an atom domain with the starting
and ending basis function indexes thereafter. N-1 lines follow where the
bond domain is defined in the following way:
Do i=2,nSets
Read(*,*)(Bonds(i,j),j=1,i-1)
Enddo
Finally a scalar is given which scales the defined bond domains.
The LoProp-functions will almost coincide with the original input AO-basis, although the localization will modify the meaning slightly, hence it is not possible to exactly localize the perturbation to a group of atoms; LOPROP is a way to come close to perfect localization. FFPT calls LOPROP internally and no call to LOPROP has to specified by the user. |
| CUMUlative | Adds the perturbation to the current H0, enabling many consecutive FFPT calls. Without this keyword, the perturbation always starts from the unperturbed H0. |
8.16.3.2 Input example
The following input will prepare the one-electron integral file generated by
SEWARD for subsequent finite-field perturbation calculations by adding
a linear electric field in z-direction.
&FFPT
DIPO
Z 0.001
Response properties are obtained by numerical differentiation of the total energy
with respect to the field parameter. For definitions of the response properties
the interested reader is referred to the paper of A.D. Buckingham in Adv.
Chem. Phys., Vol 12, p 107 (1967). According to the definition of the dipole
moment, it is obtained as the first derivative of the energy with
respect to the field strength. Similarly, the dipole polarizability is given
by the second derivative of the energy with respect to the field strength.
Next: 8.17 gateway Up: 8. Programs Previous: 8.15 falcon

