MOLCAS manual:

Next: 8.40 quater
Up: 8. Programs
Previous: 8.38 poly_aniso
Subsections
8.39 qmstat
8.39.1 Description
QmStat couples a quantum chemical region to a
statistically mechanically described surrounding thus creating
an effective Hamiltonian for the quantum chemical region
Heff.. This way solvent effects can be
accounted for.
The surrounding is discrete in contrast to the
continuum models, such as PCM (also available in MOLCAS see
Seward). The explicit representation of the solvent
enables a more accurate description of the solvation,
but also makes the model more complex and significantly
less ``black-box''.
For example, the interaction within the statistical
mechanical surrounding has to be accounted for with an
accurate enough force-field. In its present implementation
QmStat only treats water as described by an early
version of NEMO, which includes polarization of the
molecules [78]. Also, the interaction
between the quantum chemical region (typically the solute) and
the surrounding (typically the solvent) has to be considered
in more detail than in a continuum model.
Several approaches to discrete (or explicit) solvation are
thus possible.
The approach in QmStat is summarized below, see
also [79,80,81,82].
To include entropic effects to the solvation phenomena,
QmStat uses the Metropolis–Monte Carlo simulation
technique. This means that random steps are taken in the
space of solute–solvent configurations, some of which are
accepted, others rejected, on account of the usual energy
difference criteria. This implies that at each step, an
energy has to be evaluated. Using normal quantum chemical
methods, this is usually too restrictive, since roughly one
million Monte Carlo steps are required to converge the statistical
mechanical treatment. QmStat proceeds by doing
simplifications to the quantum chemistry, not the statistical
mechanics, as is the more common way forward. QmStat
is therefore a hybrid QM/MM methods (according to one
existing terminology).
Two simplified quantum chemical methods are presently available:
orbital basis Hartree–Fock and a state basis formulation, which
is approximate to the CASSCF method. Both formulations uses the
fact that there is only minor differences in the electronic
structure for the different configurations in the Monte Carlo
simulation. Therefore, a basis as general as the standard atomic
orbital basis sets is redundant. QmStat constructs
either a more compact orbital basis or a more compact basis
in terms of states to expand the solvated wave function. This
requires some work before the simulation, but has the advantage
that during the simulation, less computational work is
needed.
Finally, a comment on the way the energy is computed for a given
configuration is needed. The evaluation of the interactions between the solvent
molecules is prescribed by the construction of the force-field and
are relatively simple.
The interaction between the quantum chemical region and the
solvent is formulated to include electrostatic and non-electrostatic
interactions. The former is described in a multi-center multipole
expanded way, while the latter models the effect the antisymmetry
principle between solute and solvent electrons has on the
solute electronic structure. Its formulation is similar to
pseudo-potentials. Also a phenomenological term for the dispersion
is added. Long range electrostatics, finally, is described with a
dielectric cavity model.
8.39.2 Dependencies
The dependencies of QmStat differ for the two quantum
chemical methods. In the Hartree–Fock description, Seward,
FfPt, Scf, Averd, MpProp and
Motra typically have to precede. If an orbital basis is taken from
somewhere else FfPt, Scf and Averd
are not mandatory. For the RASSI alternative, typically
Seward, Scf, RasScf, MpProp
and Rassi precede QmStat.
8.39.3 Files
Below is a list of the files that are used/created by the program
QmStat.
| File | Contents
|
| ONEINT | One-electron integral file generated by the program SEWARD.
|
| RUNFILE | File for communication of auxiliary information generated by the program
SEWARD.
|
| RUNFILEW | File for communication of auxiliary information generated by the program
SEWARD for the solvent molecule.
|
| AVEORB | (Only for Hartree–Fock alternative). Average orbitals generated by AVERD.
If other orbitals are to
be used, they should be given the above name; in other words, the orbitals
must not be created by AVERD, it is only customary.
|
| SOLORB | Solvent orbitals generated by SCF.
|
| TRAONE | (Only for Hartree–Fock alternative). Molecular orbital transformed one-electron
integrals generated by MOTRA.
|
| TRAINT | (Only for Hartree–Fock alternative). Molecular orbital transformed two-electron
integral generated by MOTRA.
|
| MPPROP | File generated by MPPROP.
|
| DIFFPR | Exponents and Prefactors for a Slater desciption of the Electrostatics to take
into account the penetration effects due to the overlap.File generated by MPPROP.
|
| RASSIM | (Only for the RASSI alternative). The transition density matrix generated
by RASSI. The keyword TOFILE has to be given in
the input to RASSI.
|
| EIGV | (Only for the RASSI alternative). Information about the eigenvectors and
their energy generated by RASSI (TOFILE needed).
|
| ADDON* | File with additional one-electron perturbation to be added
to the Hamiltonian matrix. This file is only required if EXTERNAL
is used.
|
| File | Contents
|
| STFIL* | Start files in which solvent configurations are stored at intervals during
the simulation. They enable the simulation to restart, hence they can
also be as input to Qmstat.
|
| SAFIL* | Sampling files in which a selection of configurations are stored for
analysis. They can in some applications also act as input to Qmstat,
usually in free-energy perturbation calculations.
|
| EXTRA* | Extract files which are formatted files in which data from the analysis
of the sampling files are stored.
|
8.39.4 Input
The complexity inherit in a discrete solvent model engenders a,
potentially, complex input. To (hopefully) make the input transparent
the set of keywords are ordered in several tiers. Below all keywords and
their sub- and subsubkeywords are presented.
A keyword with several tiers should typically be of the
following form
SIMUlation
...(keywords on higher tier)
END simulation
Also consult the input example below and the examples in section
![[*]](crossref.png) for guidance. Mandatory keywords
are highlighted. for guidance. Mandatory keywords
are highlighted.
| Keyword | Meaning
|
| TITLe | Title to the calculation.
|
| SIMUlation | Keywords relating to the how the simulation is to be performed and under
which conditions.
- RADIus Initial radius of the dielectric cavity. The radius is also
specified on the startfile and has higher priority than the radius given
with the present keyword.
- PERMittivity Permittivity of the dielectric continuum. 80 on
default.
- TEMPerature Temperature in Kelvin. Default is 300.
- PRESsure Macroscopic pressure in atmosphere. Default is 1 atm.
- SURFace Surface tension parameter for the cavity. Default is
for air-water interface.
- TRANslation Maximal translation in the simulation
(in a.u. )Default is 0.0 a.u.
- ROTAtion Maximal angle for rotation of solvent around
molecular axes. Default is 0o.
- CAVIty Maximal modification of radius of dielectric cavity.
Default is 0.0 a.u.
- FORCe Force constant for the harmonic potential that presents
a bias in the simulation for configurations with the QM-region close
to the center of the cavity. Default is 0.001.
- BREPulsion Parameter for the Repulsion energy that keeps the QM-region away from the boundary. Default is 0.0 a.u.
- SEED Seed to the pseudo-random number generator.
- PARAlleltemp A parallel tempering procedure is performed to boost sampling. It is mainly used in systems with small transition elements in the Markov chain, which will give difficult samplings. Three lines follow: First line
gives the number of different temperatures to perform the simulation, NTemp. In the second line Ntemp integers are given, each of these specify a file to store the configuration for each temperature. Third line gives the NTemp temperatures used
for the tempering procedure.
- END_Simulation Parameters Marks the end of the input to the simulation parameters.
|
| THREshold | Followed by three numbers. First the threshold for the induced
dipoles in the generalized self-consistent field method for the solution
of the mutual polarization problem is specified. Second the the threshold
for the energy in the same method is given. Finally the maximum
number of iterations in the method is specified. Defaults are 0.0001 0.0000001
and 30.
|
| STEPs | Followed by two entries. Number of macrosteps and number of microsteps.
The total number of steps is the product of the two numbers above. At
the end of each macrostep the relevant STFIL is up-dated. Default
is 1 and 1.
|
| RUN | Specify type of simulation. 'QMEQ' means quantum chemical equilibration;
only the startfile is up-dated. 'QMPR' means quantum chemical
production; startfile is up-dated and sampfile constructed. Observe
that if 'QMPR' is specified a line with two entries follows in which
the interval of sampling is specified and on which sampfile (1-7) the
data is to be stored. 'ANAL' means an analysis of the stored results
is to be performed.
|
| PRINt | Print level. 1 is default and anything above this number can generate
large outputs. No higher than 10 is recommended for non-developers.
|
| EXTErnal | An external perturbation is to be added to the Hamiltonian
in the Rassi alternative. The arguments are number of perturbation
matrices, N, followed by N lines. Each line has the form: ci a scalar
with which the perturbation can be scaled, Vi is a character string with
the label of the perturbation as given on SEWARD's one-electron integral file,
nci is the component number of the perturbation.
A final expression for the perturbation would be:
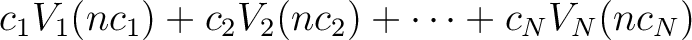 . .
|
| CONFiguration | Keywords relating to from which source the initial solvent
configuration is to be obtained. It is mandatory to
specify a source.
- ADD Followed by one number specifying how many solvent
molecules that are to be added at random to the cavity. This is the
worst way to start a simulation since it will take a lot of time to
equilibrate the system.
- FILE Signify that start configuration is to be read from
some file.
- STARtfile Read solvent configuration from startfile.
- SCRAtch Read solvent configuration from startfile and place
the QM-region as given on RUNFILE.
- COPY Read solvent and QM configuration from startfile.
This is he keyword to use if a simulation is to be restarted.
Observe that consistent startfile and RUNFILE must be used.
- CM_ _ Read solvent configuration from startfile and place
the QM in the center of mass of the QM placed on startfile.
For any of the previous keywords two numbers are given, Nin and Nout which specify from
which startfile QmStat is supposed to read and write,
respectively
- SAMPfile Read solvent configurations put on a
sampfile and analyze them. Two numbers are given, Nin and
Nextr which specify from which sampfile QmStat is
supposed to read and on which extract file the results are to
be put.
- INPUt The starting configuration is to be read from
the input. The coordinates are given after the keyword
COORdinates in the second tier to the SOLVent
keyword. One number as argument: the startfile to which
configurations are written.
- END_Configuration Marks the end of the input to the initial configuration.
|
| EDIT | Signify that a startfile is to be edited. If this keyword is
given, then no simulation will be performed.
- DELEte Two rows follow; on the first Nin and Nout
are given which specify the startfile to read from and write to,
respectively; on the second the number of solvent molecules to
delete. The solvent molecules farthest away from origin are
deleted.
- ADD The form of the arguments as DELEte
above, only the second row give number of molecules to add.
Observe that the keyword RADIus will with the
present keyword specified give the radius of the cavity of
the edited startfile.
- QMDElete Delete the QM-region and substitute it by water molecules.
One row follows with two numbers, which specify the startfile to read from and write to, respectively.
- DUMP Dump startfile coordinates in a way suitable for graphical display.
Two rows follow; on the first a character string with the format the coordinated
will be dumped; on the second Nin specifies the startfile to read.
Currently there is only one format for this keyword: MOLDen.
- END_EditStartFile Marks the end of the input to edit the startfile.
|
| QMSUrrounding | Keywords that are related to the interaction between surrounding
and the quantum chemical region.
- DPARameters
Parameters for the dispersion interaction.
Follow N lines, which N the number of atoms in the QM-region. The general form for each line is: d1 and d2 where d1 is the dispersion parameter between one atom of the QM-region and the water oxygen, and d2 is the same but regarding to the hydrogen of the water.The order of the QM atoms is given by RUNFILE.
- ELECtrostatic
Parameters to describe the electrostatic penetration using Slater integrals.
- THREsholds
Two number follow. First, the cutoff (distance Quantum Site-Classical molecule) to evaluate penetration effects. Default is 6 a.u.
Second, difference between two Slater exponents to not be consider the same value. Default is 0.001.
- NOPEnetration
No electric penetration is considered in the calculations. Penetration is considered by default.
- QUADrupoles
Electrostatic Penetration computed in quadrupoles. Default is that penetration is computed up to dipoles.
- END Electrostatic
Marks the end of the input to the electrostatic penetration computed by Slater.
- XPARameters
Parameters to describe the repulsion energy.
- S2
The parameter for the
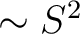 term. Default zero. term. Default zero.
- S4
The parameter for the
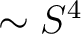 term. Default zero. term. Default zero.
- S6
The parameter for the
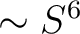 term. Default zero. term. Default zero.
- S10
The parameter for the
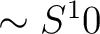 term. Default zero. term. Default zero.
- CUTOff
Two numbers follow. The first is the cut-off radius such as if
any distance from the given solvent molecule is longer than
this number, the overlap term is set to zero. The second
is a cut-off radius such as if any distance from the given
solvent molecule is shorter than this number the energy is
set to infinity, or practically speaking, this configuration is
rejected with certainty. Defaults are 10.0 a.u. and 0.0 a.u.
- END XParameters Marks the end of the input to the repulsive parameters.
- DAMPing
- DISPersion
Input parameters to a dispersion damping expression. The parameters
are number obtain from a quantum chemical calculation. All lines
have the form: Cval, Qxx, Qyy, Qzz where
Cval is the valence charge and Q** are diagonal terms
in the quadrupole tensor. First two lines are for the hydrogen
atom then the oxygen atom in a water molecule. Next follows as
many lines as atoms in the QM region. All these quantities
can be obtained from a calculation with MpProp.
The numbers are given as input so that the user can if it is found
to be needed, modify the damping. Default is no damping.
The order of the atoms in the QM region is given by RUNFILE.
- FIELd
The electric field between QM region and surrounding is damped.
Three numbers are arguments:CO, CH, N where they are
parameters to a field damping expression
(
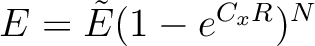 ) where x is O if the point
in the surrounding is on a oxygen atom, H if on a hydrogen
atom; R is the distance between the point in the QM region
and the points in the surrounding. ) where x is O if the point
in the surrounding is on a oxygen atom, H if on a hydrogen
atom; R is the distance between the point in the QM region
and the points in the surrounding.
- END Damping
Marks the end of the input to the Damping parameters.
- END QmSurrounding
Marks the end of the input related to the interaction between surrounding
and the quantum chemical region.
|
| SOLVent | Keywords that govern the solvent-solvent interaction and some
other initial data. Most of these numbers are presently fixed
and should not be altered.
- COORdinates
If solvent coordinates are to be given explicitly in input. First
line gives number of particles to add. Then follows three times
that number lines with coordinates for the oxygen atom and the
hydrogen atoms.
If the keyword SINGle-point has been given the
present keyword assumes a different meaning (see description
of SINGle-point).
- CAVRepulsion
Two parameters that regulate the repulsion with the boundary
of the cavity. Defaults are 30.0 and 0.06.
- ATCEchpol
Five numbers follow: number of atoms, centers, charges, polarizabilities and
slater sites. Defaults are 3, 5, 4, 3 and 5, respectively.
- CHARge
Four numbers follow: the partial charge on the hydrogen atoms
and the partial charge on the pseudo-centers.
- POLArizability
Three numbers follow: the polarizability on the oxygen atom
and on the two hydrogen atoms.
- SLATer
Magnitude of Slater Prefactors and exponents. One mumber follow: 0 is slater description of electrostatics up to charges, 1 up to dipoles.
Then it follows N times (where N is the number of Slater centers) three lines if description up to charge. First line Slater exponent
for charges, second line Slater Prefactor and third line nuclear charge of the center. If the description goes up to dipole, N times
five lines follows. First two lines are the same as charge description, third line is Slater exponent for dipole, fourth line is the
three Slater Prefactors for the dipole (one for each cartesian coordinate) and fith line is the nuclear charge of the center. Defaults: See papers of Karlstrom. If the number of Slater sites is modified this keyword should be after ATCEchpol
- END Solvent
Marks the end of the input that govern the solvent-solvent interaction.
|
| RASSisection | This section provides the information needed to perform QMSTAT calculations
using the RASSI-construction of the wave function.
- JOBFiles First number give the number of JOB-files
that was generated by RasScf (i. e. how many
RASSCF calculations that preceded QmStat). The
following numbers (as many as the number of JOB-files) specify
how many states each calculation rendered. So for example if
a State-Average (SA) RASSCF calculation is performed with two
states, the number should be 2.
- EQSTate Which state interacts with the surrounding.
Should be 1 if it is the ground state, which also is the
default.
- MOREduce A Reduction of the Molecular Orbitals is performed.
One number as argument: the threshold giving the value of the lowest
occupation number of the selected natural orbitals [81].
- CONTract The RASSI state basis are contracted.
One number as argument: the threshold giving the value of the lowest
RASSCF overlap for the RASSI state basis [81].
- LEVElshift Introduce levelshift of RASSI states. Three lines must be written.
First line gives the number of levelshifts to perform. Then follows the states
to levelshift (as many as the number of levelshifts). Finally, the value of the
levelshift for each state is given.
- CISElect The QM solvent overlap is used as the criterion to choose
the state that interacts with the surrounding. Three lines follow. One entire:
among how many states can be chosen the interacting state, N. The
second line, N entries giving the number of each state. Finally, N scaling
factors, one for each state, of the overlap.
- END RassiSection
Marks the end of the input that govern the Rassi calculations.
|
| SCFSection | This section provides additional information to perform QMSTAT calculations
using the SCF-construction of the wave function.
- ORBItals
Two numbers are required: how many orbitals that are to be used
how many occupied orbitals there are in the QM region.
as a basis in which to solve the Hartree-Fock equation, and
- END ScfSection
Marks the end of the input that govern the Scf calculations.
|
| SINGle-point | This keywords signals that a set of single point calculations
should be performed; this is typically what one needs when
fitting parameters. The keyword gives the COORdinates
keyword in the SOLVent section a new meaning. The first
row then gives the number of points in which a single-point calculation
should be performed and the coordinates that follow give the
coordinates for the water monomer. QmStat then run each
solute-monomer solvent configuration specified and the energy (among
other things) is computed. The keyword
thus overrides
the usual meaning of the input. Observe that the permittivity
has to be set to 1 if one attempts to reproduce a quantum chemical
supermolecular potential.
|
| EXTRact Section | Give details about the analysis performed to the results stored in the
sampfile.
- TOTAl energy
The total energy of the whole system is extracted.
- DIPOle
The three components and the total dipole of the QM-region are extracted.
- QUADrupole
The six components and the quadrupole of the QM-region are extracted.
- EIGEn
The Eigenvalues of the RASSI matrix and the eigenvectors are extracted.
Follow by a number and a "YES" or "NON" statement. The number gives the
highest state where the eigenvalue is extracted. YES means that also the
corresponding eigenvectors are extracted.
- EXPEctation values
The expectation values of H0 and main perturbations: Vel, Vpol and
Vn-el are extracted. If keyword EIGEn is specified it is done
for the same states as this keyword, otherwise the extraction is performed for
the equilibrated state. Observe that the expectation values are for the
final wave function of the QM-region in solution, so H0 is not the same as
for the isolated QM-region.
- ELOCal
The local expectation values of Vel and Vpol for the multipole
expansion sites are extracted. Two lines follow. First, gives for how many sites
these values will be extracted, N. Second line, N entries giving the number
of each site. If keyword EIGEn is specified the extraction is done
for the same states as this keyword, otherwise it is performed for the
equilibrated state.
- MESP
The Main Electrostatic potential, field and field gradients will
be obtained in order to produce perturbation integrals that will
be used to optimize the intramolecular geometry of the QM system.
Observe that this keyword will change the one electron integrals file,
so it is advised to make a copy of the original file.
After running this option ALASKA and SLAPAF must be running with the new one
electron integrals file in order to produce the gradients and a new geometry
in the geometry optimization procedure.
- END ExtractSection
Marks the end of the input that give details about the analysis performed.
|
The following input uses the Rassi alternative and restarts from
startfile.0 and write to startfile.1 every 1000th step, where
the total number of steps is 200*1000. A set of parameters are
given which are for an organic molecule with one carbon,
one oxygen and two hydrogen atoms. The order in the previous
SEWARD and RASSCF calculations for the atoms is carbon,
oxygen, hydrogen 1 and hydrogen 2. The dispersion is damped. Finally,
there are sixteen RASSCF calculations preceeding and the last
two are state-average since two states are collected from these
files; the ground state interacts with the surrounding.
&QmStat &End
Simulation * Simulation parameters.
Translation
0.03 * Maximun translation step of water.
Rotation
1.0 * Maximun rotation step of water.
Cavity
0.05 * Maximun variation of the cavity radius for step.
End Simulation
Steps * Number of macro and microsteps.
200 1000
Configuration * How the start configuration is readed.
Start * The cordinates are taken form a startfile.
Copy * The coordinates of the QM region are the same as in the startfile.
0 1
End Configuration
QmSurrounding
DParameters * Dispersion parameters.
35.356 4.556 * Carbon_QM-Oxygen_wat Carbon_QM-Hydrogen_wat.
16.517 2.129 * Oxygen_QM-Oxygen_wat Oxygen_QM-Hydrogen_wat.
10.904 1.405 * Hydrogen1_QM-Oxygen_wat Hydrogen1_QM-Hydrogen_wat.
10.904 1.405 * Hydrogen2_QM-Oxygen_wat Hydrogen2_QM-Hydrogen_wat.
XParameters * QM-Solvent Repulsion Parameters.
S2
-0.375
S6
1.7
End XParameters
Damping * Dispersion Damping.
Dispersion
-6.64838476 -5.22591434 -4.32517889 -4.58504467 * Water Hydrogen.
-.34146881 -0.21833165 -0.22092206 -0.21923063 * Water Oxygen.
-4.23157193 -1.91850438 -2.28125523 -1.91682521 * Quamtum Carbon.
-6.19610865 -3.90535461 -4.73256142 -3.77737447 * Quantum Oxygen.
-.57795931 -0.42899268 -0.43228880 -0.43771290 * Quantum Hydrogen 1.
-.57795931 -0.42899268 -0.43228880 -0.43771290 * Quantum Hydrogen 2.
End Damping
End QmSurrounding
RassiSection
JobFiles * Number of JobFiles.
16
1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 * One state is collected form all JobFiles
* except from the two last ones, which two
* are collected.
EqState * The state interacting with the surrounding.
1
End RassiSection
End of Input
Next: 8.40 quater
Up: 8. Programs
Previous: 8.38 poly_aniso
|


























